Filed Pursuant to Rule 424(b)(5)
Registration No. 333-208330
PROSPECTUS SUPPLEMENT
(To Prospectus dated December 22, 2015)

19,772,727 Shares of Common Stock
Warrants to Purchase 11,863,636 Shares of Common Stock
We are offering 19,772,727 shares of our common stock and warrants to purchase up to 11,863,636 shares of our common stock at an exercise price of
$1.42 per whole share of common stock. The shares of common stock and warrants will be sold in units, with each unit consisting of one share of common stock and 0.6 of a warrant to purchase one share of common stock. Each unit will be sold at a
price of $1.10 per unit. The shares of common stock and warrants will be mandatorily separable immediately upon issuance.
Our common stock is
listed on The NASDAQ Capital Market under the symbol “GALE.” On January 6, 2016 the last reported sale price of our common stock on The NASDAQ Capital Market was $1.29 per share.
The warrants are not and will not be listed for trading on The NASDAQ Capital Market, or any other securities exchange or nationally recognized trading
system. There is no market through which the warrants may be sold, and purchasers may not be able to resell the warrants purchased under this prospectus supplement. This may affect the pricing of the warrants in the secondary market, the
transparency and availability of trading prices, and the liquidity of the warrants.
Investing in our securities
involves significant risks. See “Risk Factors” beginning on page S-11 of this prospectus supplement and on page 1 of the accompanying prospectus and the documents incorporated by reference in this prospectus
supplement and the accompanying prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or
disapproved of these securities or determined if this prospectus supplement or the accompanying prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
|
|
|
|
|
|
|
|
|
| |
|
Per
Unit |
|
|
Total |
|
| Price to the public |
|
$ |
1.10 |
|
|
$ |
21,750,000 |
|
| Underwriting discounts and commissions(1) |
|
$ |
0.066 |
|
|
$ |
1,305,000 |
|
| Proceeds, before expenses, to us |
|
$ |
1.034 |
|
|
$ |
20,445,000 |
|
| (1) |
For additional information about the expenses for which we have agreed to reimburse the underwriters in connection with this offering, see “Underwriting” beginning on page S-30 of this prospectus supplement.
|
We have granted the underwriters an option for a period of 30 days from the date of this prospectus supplement to
purchase up to an additional 2,965,909 shares of common stock at a price of $1.0246 per share and/or additional warrants to purchase up to 1,779,545 shares of common stock at a price of $0.0094 per warrant to cover over-allotments, if any.
The underwriters expect to deliver the units on or about January 12, 2016.
RAYMOND JAMES
|
|
|
|
|
|
|
|
|
| Roth Capital Partners |
|
Noble Financial Capital Markets |
|
FBR |
|
|
Maxim Group LLC |
|
The date of this prospectus supplement is January 7, 2016.
TABLE OF CONTENTS
Prospectus Supplement
Prospectus
i
ABOUT THIS PROSPECTUS SUPPLEMENT
This prospectus supplement is part of the registration statement on Form S-3 (File No. 333-208330) that we filed with the Securities and
Exchange Commission, or the SEC, using a “shelf” registration process to register sales of our securities under the Securities Act of 1933, as amended, or the Securities Act. This document consists of two parts. The first part is this
prospectus supplement, including the documents incorporated by reference, which describes the specific terms of this offering. The second part is the accompanying prospectus filed with the SEC as part of the registration statement that was declared
effective by the SEC on December 22, 2015, including the documents incorporated by reference, that gives more general information, some of which may not apply to this offering. Generally, when we refer only to the “prospectus,” we are
referring to both parts combined. This prospectus supplement may add to, update or change information in the accompanying prospectus and the documents incorporated by reference into this prospectus supplement or the accompanying prospectus.
We sometimes collectively refer to the shares of common stock and warrants offered hereby and the shares of common stock underlying the
warrants as the “securities.”
If information in this prospectus supplement is inconsistent with any document incorporated by
reference that was filed with the SEC before the date of this prospectus supplement, you should rely on this prospectus supplement. This prospectus supplement, the accompanying prospectus and the documents incorporated into each by reference include
important information about us, the securities being offered and other information you should know before investing in our securities. You should also read and consider information in the documents to which we have referred you in the section of
this prospectus entitled “Where You Can Find More Information.”
You should rely only on this prospectus supplement, the
accompanying prospectus and the information incorporated or deemed to be incorporated by reference in this prospectus supplement and the accompanying prospectus. We and the underwriters have not authorized anyone to provide you with information that
is in addition to or different from that contained or incorporated by reference in this prospectus supplement and the accompanying prospectus. If anyone provides you with different or inconsistent information, you should not rely on it. This
prospectus supplement, the accompanying prospectus and any related free writing prospectus do not constitute an offer to sell or the solicitation of an offer to buy securities in any jurisdiction to any person to whom it is unlawful to make such
offer or solicitation in such jurisdiction. You should not assume that the information contained or incorporated by reference in this prospectus supplement or the accompanying prospectus is accurate as of any date other than as of the date of this
prospectus supplement or the accompanying prospectus, as the case may be, or in the case of the documents incorporated by reference, the date of such documents regardless of the time of delivery of this prospectus supplement and the accompanying
prospectus or any sale of our securities. Our business, financial condition, liquidity, results of operations and prospects may have changed since those dates.
We further note that the representations, warranties and covenants made by us in any agreement that is filed as an exhibit to any document
that is incorporated by reference into this prospectus supplement or the accompanying prospectus were made solely for the benefit of the parties to such agreement, including, in some cases, for the purpose of allocating risk among the parties to
such agreements, and should not be deemed to be a representation, warranty or covenant to you. Moreover, such representations, warranties or covenants were accurate only as of the date when made. Accordingly, such representations, warranties and
covenants should not be relied on as accurately representing the current state of our affairs.
The industry and market data contained or
incorporated by reference in this prospectus supplement and the accompanying prospectus are based either on our management’s own estimates or on independent industry publications, reports by market research firms or other published independent
sources. Unless otherwise indicated, all information contained or incorporated by reference in this prospectus supplement and the accompanying prospectus concerning our industry in general or any segment thereof, including information regarding our
general expectations and market opportunity, is based on management’s estimates using internal data, data from industry related publications, consumer research and marketing studies and other externally obtained data.
ii
PROSPECTUS SUPPLEMENT SUMMARY
This summary highlights selected information appearing elsewhere in this prospectus supplement or in the accompanying prospectus or
incorporated by reference into this prospectus supplement and the accompanying prospectus and does not contain all of the information that may be important to you or that you should consider before investing in our securities. Before making an
investment decision, you should read this prospectus supplement, the accompanying prospectus and the information incorporated by reference in this prospectus supplement and the accompanying prospectus in their entirety, including “Risk
Factors” beginning on page S-12 of this prospectus supplement.
About Galena
Overview
Galena Biopharma, Inc.
(“we,” “us,” “our,” “Galena” or the “company”) is a biopharmaceutical company committed to the development and commercialization of targeted oncology therapeutics that address major unmet medical
needs. Galena’s development portfolio is focused primarily on addressing the rapidly growing patient populations of cancer survivors by harnessing the power of the immune system to prevent cancer recurrence. The Company’s pipeline consists
of multiple mid- to late-stage clinical assets, including novel cancer immunotherapy programs led by NeuVax™ (nelipepimut-S), GALE-301 and GALE-302. NeuVax is currently in a pivotal, Phase 3 clinical trial with several concurrent Phase 2 trials
ongoing both as a single agent and in combination with other therapies. GALE-301 is in a Phase 2a clinical trial in ovarian and endometrial cancers and in a Phase 1b clinical trial given sequentially with GALE-302.
We are seeking to build value for shareholders through pursuit of the following objectives:
| |
• |
|
Develop novel cancer immunotherapies to address unmet medical needs through the use of peptide-based vaccines targeting well-established tumor antigens. One of our key strategies is to target the adjuvant, minimum
residual disease setting, in high risk patients who are more likely to benefit from treatment via immunotherapy. Our immunotherapy programs are currently targeting two key areas: secondary prevention intended to significantly decrease the risk of
disease recurrence in breast cancer, gastric cancer, endometrial and ovarian cancers; and primary prevention intended to treat breast cancer earlier in the treatment spectrum. |
| |
• |
|
Expand our development pipeline by enhancing the clinical and geographic footprint of our technologies. We intend to accomplish this through the initiation of new clinical trials as well as through acquisition of
additional development stage products in related oncology indications. |
| |
• |
|
Leverage valuable partnerships and collaborations, as well as investigator-sponsored clinical trials, to maximize the scope of potential clinical opportunities in a cost effective and efficient manner.
|
| |
• |
|
Focus our resources on our expanding clinical development programs. On November 19, 2015 and December 24, 2015, respectively, we sold our Abstral® (fentanyl) Sublingual Tablets product and our Zuplenz®
(ondansetron) Oral Soluble Film product and related assets, and as of December 31, 2015, we ceased our commercial operations. |
S-1
The chart below summarizes the current status of our clinical development pipeline:

Novel Cancer Immunotherapies
Our targeted cancer immunotherapy approach is currently based upon two key areas: preventing secondary recurrence of cancer, which is becoming
increasingly important as the number of cancer survivors continues to grow; and, primary prevention intended to treat breast cancer earlier in the treatment spectrum. Once a patient’s tumor becomes metastatic, the outcome is often fatal, making
the prevention of recurrence a potentially critical component of overall patient care. Our programs primarily target patients in the adjuvant (after-surgery) setting who have relatively healthy immune systems, but may still have residual disease.
Minimal residual disease, or single cancer cells (occult cancer cells) or micrometastasis, that are undetectable by current radiographic scanning technologies, can result in disease recurrence.
Our therapies utilize an immunodominant peptide combined with the immune adjuvant, recombinant human granulocyte macrophage-colony stimulating
factor (rhGM-CSF), and work by harnessing the patient’s own immune system to seek out and attack any residual cancer cells. Using peptide immunogens has many potential clinical advantages, including a favorable safety profile, since these drugs
may lack the toxicities typical of most cancer therapies. They also have the potential to evoke long-lasting protection through activation of the immune system and a convenient, intradermal mode of delivery. We are currently engaged in multiple
clinical trials with NeuVax™ (nelipepimut-S), GALE-301, and GALE-302, targeting the prevention of recurrence in breast, gastric, ovarian and endometrial cancers.
NeuVax™ (nelipepimut-S)
NeuVax™ (nelipepimut-S), our lead product candidate, is a cancer immunotherapy targeting human epidermal growth factor receptor (HER2)
expressing cancers. NeuVax is the immunodominant nonapeptide derived from the extracellular domain of the HER2 protein, a well-established and validated target for therapeutic intervention in breast and gastric carcinomas. The NeuVax vaccine is
combined with GM-CSF for injection under the skin, or intradermal administration. Data has shown that an increased presence of circulating tumor cells (CTCs) may predict Disease Free Survival (DFS) and Overall Survival (OS)—suggesting a
dormancy of isolated micrometastases, which, over time, may lead to recurrence. After binding to the specific HLA molecules on antigen presenting cells, the nelipepimut-S sequence stimulates specific cytotoxic T lymphocyte (CTLs). These activated
CTLs recognize, neutralize and destroy, through cell lysis, HER2 expressing cancer cells, including occult cancer cells and micrometastatic foci. The nelipepimut immune response can also generate CTLs to other immunogenic peptides through inter- and
intra-antigenic epitope spreading.
Breast Cancer:
According to the National Cancer Institute, over 230,000 women in the U.S. are diagnosed with breast cancer annually. While improved diagnostics and targeted therapies have decreased breast cancer mortality in the U.S., metastatic breast cancer
remains incurable. Approximately 75% of breast cancer patients have tissue test positive for some increased amount of the HER2 receptor, which is associated with disease progression and decreased survival. Only approximately 20% to 30% of all breast
S-2
cancer patients—those with HER2 immunohistochemistry (IHC) 3+ disease, or IHC 2+ and fluorescence in situ hybridization (FISH) positive—have a HER2 directed, approved treatment option
available. This leaves the majority of breast cancer patients with low-to-intermediate HER2 IHC 1+/2+ ineligible for therapy and without an effective targeted treatment option to prevent cancer recurrence.
We have multiple trials currently ongoing for NeuVax. For our pivotal, Phase 3 PRESENT (Prevention of Recurrence in Early-Stage, Node-
Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) trial, NeuVax is targeting the 30,000-40,000 of the over 230,000 female breast cancer patients annually diagnosed in the U.S. who are at a higher risk of their
breast cancer recurring, which we refer to as “disease recurrence,” after achieving “no evidence of disease” (NED) status, (or becoming a “survivor”) with standard-of-care therapy (surgery, chemotherapy, radiation).
These high-risk patients have a particular molecular signature and disease status: HER2 IHC 1+/2+ (oncoprotein associated with aggressive tumor growth), node positive (disease present in the axillary lymph nodes prior to surgery), and HLA A2/A3
(human leukocyte antigen from A2/A3 patients who have the same loci of genes which represents approximately 65% of the population). Up to 25% of resectable, node-positive breast cancer patients, having no radiographic evidence of disease following
surgery and adjuvant chemo/radiation therapy, are expected to relapse within three years following diagnosis. The prognosis upon recurrence is very poor. These cancer patients presumably still had isolated, undetected tumor CTCs that led to a
recurrence of cancer in the breast (local recurrence) or in another location (metastatic disease). In addition to our Phase 3 trial, we currently have two additional Phase 2 breast cancer trials ongoing with NeuVax in combination with trastuzumab
(Herceptin®; Genentech/Roche) targeting the prevention of recurrence in expanded indications.
We also recently announced our intent
to initiate a Phase 2 trial with NeuVax as a single agent in patients with ductal carcinoma in situ (DCIS) in collaboration with the National Cancer Institute (NCI), potentially positioning NeuVax earlier in the treatment cycle towards primary
prevention. The trial will have an immunological endpoint evaluating NeuVax peptide-specific cytotoxic T lymphocyte (CTL; CD8+ T cell) response in vaccinated patients. DCIS, is defined by the NCI as a noninvasive condition in which abnormal cells
are found in the lining of a breast duct, and is the most common type of breast cancer. The abnormal cells have not spread outside the duct to other tissues in the breast. In some cases, DCIS may become invasive cancer and spread to other tissues,
and at this time, there is no way to know which lesions could become invasive. Current treatment options for DCIS include breast- conserving surgery and radiation therapy with or without tamoxifen, breast-conserving surgery without radiation
therapy, or total mastectomy with or without tamoxifen. According to the American Cancer Society, in 2015 there were an estimated 50,040 diagnoses of DCIS.
Gastric Cancer: Gastric cancer (also known as stomach cancer) is a disease in which the cells forming the inner lining of the stomach
become abnormal and start to divide uncontrollably, forming a cancerous tumor mass. Cancer can develop in any of the five sections of the stomach. Symptoms and outcomes of the disease will vary depending on the location of the cancer. Stomach cancer
is one of the leading causes of cancer deaths in several areas of the world, most notably in Asia. Annually, almost one million people will be diagnosed worldwide with stomach cancer and over 700,000 will die from the disease. More than 90% of
stomach cancers are caused by adenocarcinomas, malignant cancers that originate in glandular tissues. Overexpression of the HER2 receptor occurs in approximately 20% of gastric and gastro-esophageal junction adenocarcinomas, predominantly those of
the intestinal type. Overall, without regard to the stage of cancer, only approximately 28% of patients with stomach cancer live at least five years following diagnosis and new adjuvant treatments are needed to prevent disease recurrence.
We currently have a number of ongoing or planned clinical trials designed to expand the clinical and geographical footprint of NeuVax:
| |
• |
|
Phase 3 Ongoing: Our Phase 3 PRESENT (Prevention of Recurrence in Early- Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) study
targeted enrollment of 700 HER2 1+/2+ patients under a Special Protocol Assessment (SPA) granted by the U.S. Food and Drug Administration (FDA). The multinational, multicenter, randomized, double-blinded PRESENT trial is ongoing in North America,
Western and Eastern Europe, and Israel. The trial is fully enrolled with 758 patients. |
| |
• |
|
Phase 2b Ongoing: A randomized, multicenter, investigator-sponsored, 300 patient Phase 2b clinical trial is enrolling HER2 1+/2+ node-positive and high-risk node-negative breast cancer patients who are HLA A2+, A3+,
A24+ or A26+ to study NeuVax in combination with trastuzumab in the adjuvant setting. This trial is co-funded by Genentech/Roche (providing both trastuzumab and monetary support) and Galena (providing NeuVax and monetary support). |
| |
• |
|
Phase 2 Ongoing: An investigator-sponsored trial is ongoing to study NeuVax in combination with trastuzumab. The study will enroll 100 node positive and negative HER2 IHC 3+ patients or HER2 gene-amplified breast cancer
patients who are HLA A2+ or HLA A3+ and are determined to be at high-risk for recurrence. Partial funding for this trial comes from the Department of Defense (DoD) through the Congressionally Directed Medical Research Program via legislation known
as the Defense Appropriations Act. The grant was awarded under a Breast Cancer Research Program with the Breakthrough Award given to the lead investigator for the trial. |
S-3
| |
• |
|
Phase 2 Planned: A clinical trial, entitled, VADIS: Phase 2 trial of the Nelipepimut-S Peptide VAccine in Women with DCIS of the Breast is planned to initiate by early 2016. The Phase 2 trial will be a single-blind,
double arm, randomized, controlled trial in pre- or post-menopausal patients with DCIS and are HLA-A2 positive. VADIS will be co-funded and run in collaboration with the National Cancer Institute (NCI). |
| |
• |
|
Phase 2 Planned: A Phase 2 clinical trial in patients with gastric cancer is expected to initiate in 2016. The trial will be run in India by our partner, Dr. Reddy’s Laboratories, Ltd., as part of our NeuVax
commercialization agreement in that region with Dr. Reddy’s. |
GALE-301 and GALE-302
Our second immunotherapy franchise targets folate binding protein receptor-alpha, a well-validated therapeutic target, which has been shown to
be highly over-expressed (20-80 fold) in ovarian, endometrial and breast cancers. Both GALE-301 (E39) and GALE-302 (E39’) are immunogenic peptides that can stimulate CTLs to recognize and destroy FBP-expressing cancer cells. GALE-301 consists
of the FBP peptide E39 combined with GM-CSF, and is currently in a Phase 2a clinical trial for the prevention of recurrence in patients with ovarian and endometrial cancers. GALE-302 is an attenuated version of the E39 peptide and is currently in a
Phase 1b randomized, single-center trial investigating a novel vaccination series using GALE-301 and GALE-302 to evaluate the immune response and monitor long-term immunity. Current treatments after surgery for these diseases are principally with
platinum based chemotherapeutic agents and patients suffer a high recurrence rate; and, most patients relapse with an extremely poor prognosis. Although not powered for efficacy, promising preliminary results from the Phase 2a clinical trial of
GALE-301 were presented in September 2015 at the European Cancer Congress and demonstrated statistically significant data with the estimate for disease free survival at two years at 85.7% (1000 mcg dose group) vs. 33.6% for the control group (p <
.02), and that GALE-301 was well-tolerated with primarily Grade 1 and 2 toxicities and elicited a strong in vivo immune response.
In
November 2015, we presented preliminary data at the Society for Immunotherapy of Cancer Conference on the primary vaccine series (PVS) from a randomized Phase lb trial with GALE-301 and GALE-302 demonstrating that the in vivo immune response is
enhanced with the use of the attenuated E39’ (GALE-302) after E39 (GALE-301). Both agents were shown to be immunogenic and well tolerated with no differences in toxicities between primary vaccine sequences.
Ovarian and Endometrial Cancer: According to the NCI Surveillance, Epidemiology, and End Results (SEER) Program, new cases of ovarian
cancer occur at an annual rate of 12.1 per 100,000 women in the U.S., with an estimated 21,290 cases for 2015. Although ovarian cancer represents about 1.3% of all cancers, it represents about 2.4% of all cancer deaths, or an estimated 14,180
deaths in 2015. Approximately 1.3% of women will be diagnosed with ovarian cancer at some point during their lifetime (2010 – 2012 data). The prevalence of ovarian cancer in the U.S. is about 192,000 women, and the five-year survivorship for
women with ovarian cancer is 45.6%. Due to the lack of specific symptoms, the majority of ovarian cancer patients are diagnosed at later stages of the disease, with an estimated 75% of women presenting with advanced-stage (III or IV) disease. These
patients have their tumors routinely surgically debulked to minimal residual disease, and then are treated with platinum- and/or taxane-based chemotherapy. While many patients respond to this treatment regimen and become clinically free-of-disease,
the majority of these patients will relapse. Depending upon their level of residual disease, the risk for recurrence after completion of primary therapy ranges from 60% to 85%. Unfortunately for these women, once the disease recurs, treatment
options are limited and the disease remains incurable.
According to the NCI SEER Program, new cases of endometrial cancer occur at an
annual rate of 25.1 per 100,000 women in the U.S., with an estimated 54,870 cases for 2015. Although endometrial cancer represents about 3.3% of all cancers, it represents about 1.7% of all cancer deaths, or an estimated 10,170 deaths in 2015.
Approximately 2.8% of women will be diagnosed with endometrial cancer at some point during their lifetime (2010 – 2012 data). The prevalence of endometrial cancer in the U.S. is about 620,000 women, and the five-year survivorship for women with
endometrial cancer is 81.7%.
Hematology
GALE-401 (anagrelide controlled release (CR))
In January 2014, we announced the acquisition of the worldwide rights to anagrelide controlled release (CR), which we renamed GALE-401, through
our acquisition of Mills Pharmaceuticals, LLC. GALE-401 contains the active ingredient anagrelide, an FDA-approved product, for the treatment of patients with myeloproliferative neoplasms (MPNs) to lower abnormally elevated platelet levels. The
currently available immediate release (IR) version of anagrelide causes adverse events that are believed to be dose and plasma concentration dependent. Therefore, reducing the maximum concentration (Cmax) is hypothesized to reduce the side effects,
but preserve efficacy.
S-4
Multiple Phase 1 studies in 98 healthy subjects have shown GALE-401 reduces the Cmax of
anagrelide following oral administration, appears to be well-tolerated at the doses administered, and to be capable of reducing platelet levels. The Phase 1 program provided the desired PK/PD (pharmacokinetic/pharmacodynamic) profile to enable the
initiation of the ongoing Phase 2 proof-of-concept trial. The Phase 2, open label, single arm, proof-of-concept trial enrolled 18 patients in the United States for the treatment of thrombocytosis, or elevated platelet counts in patients with MPNs.
Phase 2 top-line safety and efficacy data was presented in June 2015 at the European Hematology Association 20th Congress, and we expect to present a more mature data set at the 57th American Society of Hematology Annual Meeting in December 2015.
Based on a regulatory meeting with the FDA, Galena believes a 505(b)(2) regulatory filing is an acceptable pathway for development and potential approval of GALE-401.
Myeloproliferative neoplasms: MPNs are a closely related group of hematological malignancies in which the bone marrow cells that produce the
body’s blood cells develop and function abnormally. The main MPNs are Polycythemia Vera (PV), Essential Thrombocythemia (ET), Primary Myelofibrosis (PMF), and Chronic Myelogenous Leukemia (CML), all of which are associated with high platelet
counts. The MPNs are progressive blood cancers that can strike anyone at any age, and for which there is no known cure.
Alliance Partners in
Therapeutic Areas
We are actively looking to leverage our technology platforms by seeking to work with pharmaceutical and
biotechnology partners in a number of therapeutic areas in oncology. Our team has experience targeting products in multiple indications, and based on this experience, we believe we can expand the clinical utility of our current development
candidates as well as discover more drug candidates by working with partners than we can develop with our own resources. We are seeking to work with partners in the discovery and development of drugs in a number of therapeutic areas and technology
platforms.
Intellectual Property
Patents and other intellectual property rights are crucial to our success. It is our policy to protect our intellectual property rights through
available means, including filing and prosecuting patent applications in the U.S. and other countries, protecting trade secrets, and utilizing regulatory protections such as data exclusivity. We also include restrictions regarding use and disclosure
of our proprietary information in our contracts with third parties, and utilize customary confidentiality agreements with our employees, consultants, clinical investigators and scientific advisors to protect our confidential information and
know-how. Together with our licensors, we also rely on trade secrets to protect our combined technology especially where we do not believe patent protection is appropriate or obtainable. It is our policy to operate without infringing on, or
misappropriating, the proprietary rights of others. The following chart summarizes our intellectual property rights:
|
|
|
|
|
|
|
|
|
| Drug Candidate |
|
Indication |
|
Scope |
|
Strategic Partner |
|
Estimated
Exclusivity
Period |
| NeuVax™ (nelipepimut-S) |
|
Breast cancer recurrence |
|
Pending and/or issued |
|
University of
Texas/MDACC/
Henry M. Jackson
Foundation |
|
2028 |
|
|
|
|
|
| NeuVax™ (nelipepimut-S) |
|
Gastric |
|
Pending and/or issued |
|
Dr. Reddy’s
Laboratories |
|
2028 |
|
|
|
|
|
| NeuVax™ (nelipepimut-S) |
|
DCIS |
|
Pending and/or issued |
|
National Cancer
Institute/
University of
Texas, MD
Anderson Cancer
Center |
|
2028 |
|
|
|
|
|
| NeuVax™ in combination with Herceptin® |
|
Breast cancer |
|
Pending and/or issued |
|
Henry M. Jackson
Foundation,
Genentech/Roche |
|
2026 |
|
|
|
|
|
| NeuVax™ in combination with other compounds |
|
Breast cancer |
|
Pending and/or issued |
|
Pending |
|
2037 |
|
|
|
|
|
| GALE-301 & GALE-302 |
|
Breast, ovarian and endometrial cancer |
|
Pending and/or issued |
|
Henry M. Jackson
Foundation |
|
2035 |
|
|
|
|
|
| GALE-401 (Anagrelide Controlled Release) |
|
Platelet Lowering |
|
Pending and/or issued |
|
BioVascular, Inc. |
|
2029 |
S-5
Out-License Agreements
Teva Pharmaceuticals
Effective December 3, 2012, we entered into a license and supply agreement with ABIC Marketing Limited, a subsidiary of Teva
Pharmaceuticals (“ABIC”). Under the agreement, we granted ABIC exclusive rights to seek marketing approval in Israel for our NeuVax product candidate for the treatment of breast cancer following its approval by the FDA or the European
Medicines Agency, and to market, sell and distribute NeuVax in Israel assuming such approval is obtained. ABIC’s rights also include a right of first refusal in Israel for all future indications for which NeuVax may be approved.
Under the license and supply agreement, ABIC will assume responsibility for regulatory registration of NeuVax in Israel, provide financial
support for local development, and commercialize the product in the region in exchange for making royalty payments to us based on future sales of NeuVax. ABIC also agrees in the license and supply agreement to purchase all supplies of NeuVax from us
at a price determined according to a specified formula.
Dr. Reddy’s Laboratories Ltd.
Effective January 14, 2014, we entered into a strategic development and commercialization partnership with Dr. Reddy’s
Laboratories Ltd. (“Dr. Reddy’s”), under which we licensed commercial rights in India to Dr. Reddy’s for NeuVax in breast and gastric cancers. Under the agreement, Dr. Reddy’s will lead the Phase 2 development of
NeuVax in India in gastric cancer, significantly expanding the potential patient population addressable with NeuVax.
Kwangdong
Pharmaceutical Co., Ltd.
Effective April 30, 2009, we entered into a license agreement with Kwangdong Pharmaceutical Co, Ltd
(Kwangdong). Under the agreement, we granted Kwangdong exclusive rights to seek marketing approval in The Republic of Korea (South Korea) for our NeuVax product candidate for the treatment of breast cancer following its approval by the FDA or the
European Medicines Agency, and to market, sell and distribute NeuVax in South Korea assuming such approval is obtained.
Recent Developments (in
reverse chronological order)
Strategic Review of Commercial Operations – Following a strategic review of our
commercial operations, we recently sold our commercial assets.
During the three months ended September 30, 2015, we completed
a strategic review of the commercial business and operations. As a result of the review, we recently sold our Abstral® (fentanyl) Sublingual Tablets and Zuplenz® (odansetron) Oral Soluble Film products and related assets as described below.
On November 19, 2015, we entered into an asset purchase agreement with Sentynl Therapeutics Inc., or Sentynl, pursuant to which we
sold to Sentynl our Abstral® (fentanyl) Sublingual Tablets product and related assets. The assets included our rights in the asset purchase agreement and the license agreement between us and Orexo AB by which we acquired rights in Abstral®.
Our future obligations under the agreements with Orexo AB have been assumed by Sentynl, except that we will continue to be responsible for certain channel liabilities related to Abstral for a specified period of time post-closing. We also will be
responsible for any pre-closing liabilities and obligations related to Abstral and have agreed in the asset purchase agreement to indemnify Sentynl for any breach of our representations, warranties and covenants in the asset purchase agreement up to
a certain agreed upon amount.
The total potential consideration payable to us under the purchase agreement is $12 million, comprised of
an $8 million upfront payment and up to an aggregate of $4 million in future payments based on Sentynl’s achievment of specified net sales of Abstral®.
On December 17, 2015, we entered into an asset purchase agreement with Midatech Pharma PLC, or Midatech, pursuant to which we sold to
Midatech on December 24, 2015 our Zuplenz® (ondansetron) Oral Soluble Film product and related assets. The
S-6
assets included our rights in the license and supply agreement between us and MonoSol Rx, LLC, or MonoSol, by which we acquired rights in Zuplenz. Our future obligations under the MonoSol
agreement have been assumed by Midatech, except that we will continue to be responsible for certain channel liabilities related to Zuplenz for a specific period of time post-closing. We also will be responsible for any pre-closing liabilities and
obligations related to Zuplenz and have agreed in the asset purchase agreement to indemnify Midatech for any breach of our representations, warranties and covenants in the asset purchase agreement up to a certain agreed upon amount.
The total potential consideration payable to us under the purchase agreement is $29.75 million, comprised of an $3.75 million upfront payment
and up to an aggregate of $26 million in future payments based on Midatech’s achievement of specified net sales of Zuplenz®.
In
a separate agreement with MonoSol entered into on December 16, 2015, we and MonSol agreed to amend the MonoSol license and supply agreement in order to reduce the number of field representatives that we were required to maintain with respect to
Zuplenz, and we agreed to pay MonoSol $900,000 of the upfront fee and 20% of any future payments we receive under the Midatech purchase agreement.
In connection with our sale of Abstral, Zuplenz and related assets, we discontinued our commercial operations as of December 31,2015.
Governmental Investigations - We have been subpoenaed in connection with federal investigations.
A federal investigation of two of the high-prescribing physicians of our former Abstral product has resulted in a criminal prosecution of the
two physicians for alleged violations of the federal False Claims Act and other federal statutes. The criminal trial of the two physicians is set for some time in 2016. We have received a trial subpoena for documents and we have been in contact with
the U.S. Attorney’s Office for the Southern District of Alabama, which is handling the criminal trial, and are cooperating in the production of documents. We are not a target or subject of that investigation. There also have been federal and
state investigations of a company that has a product that competes with Abstral in the same therapeutic class, and we have learned that the FDA and other governmental agencies may be investigating our Abstral promotion practices. On
December 16, 2015, we received a subpoena issued by the U.S. Attorney’s Office in District of New Jersey requesting the production of a broad range of documents pertaining to our marketing and promotional practices for Abstral. We have
been in contact with the U.S. Attorney’s Office for the District of New Jersey and are cooperating in the production of the requested documents.
Shareholder Litigation Settlements – We have reached agreements in principle to settle our shareholder
derivative and class action litigations.
On December 3, 2015, we reached an agreement in principle to
settle the consolidated shareholder derivative action, In re Galena Biopharma, Inc. Derivative Litigation, Civil Action No. 3:14-cv-00382-SI, pending in the United States District Court for the District of Oregon against us and certain
of our current and former officers and directors. The settlement will not become effective until approved by the Court. The settlement includes a payment of $15 million in cash by our insurance carriers, which we will use to fund a portion of the
class action settlement, and cancellation of 1,200,000 outstanding director stock options. The settlement also will require that we adopt and implement certain corporate governance measures and will provide that the plaintiffs’ counsel may
apply to the court for an award of attorneys’ fees and expenses up to $5 million, which if awarded by the court to the plaintiffs’ counsel will be paid by our insurance carriers. The settlement will not include any admission of wrongdoing
or liability on the part of us or the individual defendants and will include a full release of us and the current and former officers and directors in connection with the allegations made in the consolidated federal derivative actions and state
court derivative actions.
On December 3, 2015, we also agreed in principal to resolve and settle the securities
putative class action lawsuit, In re Galena Biopharma, Inc. Securities Litigation, Civil Action No. 3:14-cv-00367-SI, pending against us, certain of our current and former officers and directors and other defendants in the United States
District Court for the District of Oregon. The agreement, which is subject to shareholder notice and Court approval, provides for a settlement payment of $20 million to the class and the dismissal of all claims against us and the other defendants in
connection with the consolidated federal securities class actions. Of the $20 million settlement payment to the class, $16.7 million will be paid by our insurance carriers and $3.3 million will be paid by us through a combination of $2.3 million in
cash and $1 million in shares of our common stock. We will be responsible for defense costs and any settlements or judgments incurred for any related opt-out lawsuits.
S-7
GALE-302 Immunological Data Optimizing GALE-301 Presented - We presented
preliminary immunologic data for our GALE-301 and GALE-302 Phase 1b clinical trial at the Society for Immunotherapy of Cancer (SITC) 30th Anniversary Annual Meeting.
On November 7, 2015 we presented the poster, entitled, “Preliminary report of a clinical trial supporting the sequential use
of an attenuated E39 peptide (E39’) to optimize the immunologic response to the FBP (E39+GM-CSF) vaccine,” that compared three primary vaccine series (PVS) sequences of GALE-301 (E39) and GALE-302 (E39’) in ovarian and breast cancer
patients to optimize the ex vivo immune responses, local reactions (LR), and delayed type hypersensitivity (DTH) reactions. The data demonstrated that the in vivo immune response is enhanced with the use of the attenuated E39’
(GALE-302) after E39 (GALE-301). The optimal vaccination sequence utilizing three inoculations of GALE-301 followed by three inoculations of GALE-302 produced the most prominent and statistically significant LR and DTH responses.
Patent Infringement Litigation Settlement – We settled the patent infringement litigation involving Abstral.
On October 27, 2015, we reported that on October 23, 2015 we, Orexo AB and Actavis Laboratories Fl, Inc. (“Actavis”)
had entered into a settlement and license agreement, which will permit Actavis to enter the market with a generic or authorized generic version of Abstral in June 2018 or earlier under certain circumstances. The litigation, Orexo AB v. Actavis
Laboratories FL, Inc., CA No. 3:15-cv-00826, was dismissed with prejudice by the United States District Court for the District of New Jersey, and the settlement and license agreement has been filed with the Federal Trade Commission and the
Department of Justice under applicable law. This settlement and license agreement was assigned to Sentynl pursuant to the asset purchase agreement described above related to our sale of Abstral.
Collaboration with the National Cancer Institute – We announced a Phase 2 clinical trial with NeuVax in Ductal Carcinoma in
Situ Patients.
On September 30, 2015, we announced a collaboration with the National Cancer Institute (NCI) to initiate a
Phase 2 clinical trial with NeuVax in patients diagnosed with Ductal Carcinoma in Situ (DCIS). The trial will be entitled VADIS: Phase 2 trial of the Nelipepimut-S Peptide Vaccine in Women with DCIS of the Breast. The University of Texas M.D.
Anderson Cancer Center (MDACC) Phase I and II Chemoprevention Consortium will be the lead clinical trial site for this multi-center trial with Elizabeth Mittendorf, M.D., Ph.D. serving as the study Principal Investigator. The Consortium is funded
through the Division of Cancer Prevention at NCI, which will provide financial and administrative support for the trial. We will provide NeuVax, as well as additional financial and administrative support. The single-blind, double arm, randomized,
controlled trial is expected to be initiated in the first quarter of 2016.
Financial Condition
We had cash and cash equivalents of approximately $34.8 million as of September 30, 2015. We believe that our existing cash and cash
equivalents together with the net proceeds of this offering should be sufficient to fund our operations for at least one year. This projection is based on our current planned operations and revenue expectations and is subject to changes in our plans
and uncertainties inherent in our business, and we may need to seek to replenish our existing cash and cash equivalents sooner than we project. We also have funding available under our purchase agreement with Lincoln Park Capital Fund, LLC (subject
to the lock-up restrictions described in the “Underwriting” section of this prospectus supplement) and sales agreements with MLV & Co. and Maxim Group LLC described in the accompanying prospectus, but there is no guarantee that
such funding will be available to us on favorable terms or will be sufficient to meet all of our future funding needs.
Corporate Information
Our principal executive offices are located at 2000 Crow Canyon Place, Suite 380, San Ramon, California 94583, and our phone number is
(855) 855-4253. Our website address is www.galenabiopharma.com. We do not incorporate the information on our website into this prospectus, and you should not consider such information part of this prospectus.
We were incorporated as Argonaut Pharmaceuticals, Inc. in Delaware on April 3, 2006 and changed our name to RXi Pharmaceuticals
Corporation on November 28, 2006. On September 26, 2011, we changed our company name from RXi Pharmaceuticals Corporation to Galena Biopharma, Inc.
S-8
The Offering
|
|
|
| Common stock offered by us pursuant to this prospectus supplement |
|
19,772,727 shares (excluding 11,863,636 shares of common stock issuable upon exercise of the warrants being offered in this offering). This prospectus supplement also relates to the offer and sale of the shares of common stock
underlying the warrants being offered by us. |
|
|
| Warrants offered by us |
|
Warrants to purchase up to 11,863,636 shares of our common stock at an exercise price of $1.42 per whole share. The warrants will be exercisable upon issuance and will expire on the five-year anniversary of issuance. See
“Description of Our Securities.” |
|
|
| Common stock to be outstanding after this offering |
|
181,673,173 shares, or 193,536,809 shares of our common stock assuming the warrants offered in this offering were to be immediately issued and exercised in full. |
|
|
| Over-allotment option |
|
We have granted the underwriters a 30-day option to purchase up to 2,965,909 additional shares of common stock at a price of $1.0246 per share and/or additional warrants to purchase up to 1,779,545 shares of common stock at a price
of $0.0094 per warrant to cover over-allotments, if any. |
|
|
| Use of proceeds |
|
We currently intend to use the net proceeds from this offering to fund our Phase 3 PRESENT study of NeuVax and other clinical trials of our product candidates, and to augment our working capital and our general corporate purposes.
See “Use of Proceeds” on page S-25 of this prospectus supplement for more information about our intended use of the net proceeds. |
|
|
| Dividend policy |
|
We do not anticipate paying any cash dividends on our common stock. |
|
|
| The NASDAQ Capital Market symbol |
|
Our common stock is listed on The NASDAQ Capital Market under the symbol “GALE.” The warrants are not and will not be listed on The NASDAQ Capital Market or other securities exchange or nationally recognized trading
system. |
|
|
| Risk factors |
|
Investing in our securities involves significant risks. See “Risk Factors” beginning on page S-11 of this prospectus supplement and on page 1 of the accompanying prospectus and the documents incorporated by reference in
this prospectus supplement and the accompanying prospectus. |
The number of shares of common stock shown above to be outstanding after this offering is based on 161,900,446
shares outstanding as of September 30, 2015 and excludes as of such date:
| |
• |
|
675,000 shares held in treasury; |
| |
• |
|
11,188,443 shares of our common stock subject to outstanding options having a weighted-average exercise price of $2.83 per share; |
| |
• |
|
10,250,759 shares of our common stock reserved for issuance in connection with future awards under our 2007 stock incentive plan; |
| |
• |
|
528,131 shares of our common stock reserved for future sale under our employee stock purchase plan; |
S-9
| |
• |
|
23,308,475 shares of our common stock subject to outstanding warrants having a weighted-average exercise price of $2.10 per share; and |
| |
• |
|
11,863,636 shares of common stock issuable upon exercise of warrants to be issued in this offering. |
The shares of common stock issuable upon the exercise of our outstanding warrants and the exercise price of the warrants are subject to
anti-dilution adjustments in certain circumstances. See “Dilution” for more information about these possible anti-dilution adjustments.
Unless otherwise indicated, the information contained in this prospectus supplement assumes no exercise by the underwriters of their option to
purchase up to an additional 2,965,909 shares of common stock and/or additional warrants to purchase up to 1,779,545 shares of common stock to cover over-allotments, if any.
S-10
RISK FACTORS
Investing in our securities involves significant risks. Before making an investment decision, you should carefully consider the risks
described below, together with the information under “Risk Factors” in our most recent Annual Report on Form 10-K and subsequent Quarterly Reports on Form 10-Q and the other information incorporated by reference in this prospectus. Some of
these factors relate principally to our business and the industry in which we operate. Other factors relate principally to your investment in our common stock. If any of these risks were to occur, our business, financial condition, results of
operations, cash flows or prospects could be materially and adversely affected. In such case, you may lose all or part of your investment.
The risks and uncertainties described below and in our most recent Annual Report on Form 10-K and subsequent Quarterly Reports on Form 10-Q
are not the only ones facing us. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also materially and adversely affect our business and operations.
Risks Relating to Our Former Commercial Operations
We are subject to U.S. federal and state health care fraud and abuse and false claims laws and regulations, and we recently have been subpoenaed in
connection with marketing and promotional practices related to Abstral. Prosecutions under such laws have increased in recent years and we may become subject to such prosecutions or related litigation under these laws. If we have not fully complied
with such laws, we could face substantial penalties.
Our former commercial operations are subject to various U.S. federal and
state fraud and abuse laws, including, without limitation, the federal False Claims Act, federal Anti-Kickback Statute, and the federal Sunshine Act.
A federal investigation of two of the high-prescribing physicians for Abstral has resulted in the criminal prosecution of the two physicians
for alleged violations of the federal False Claims Act and other federal statutes. The criminal trial is set for some time in 2016. We have received a trial subpoena for documents and we have been in contact with the U.S. Attorney’s Office for
the Southern District of Alabama, which is handling the criminal trial, and are cooperating in the production of documents. We do not believe we are a target or subject of that investigation. There also have been federal and state investigations of
a company that has a product that competes with Abstral in the same therapeutic class, and we have learned that the FDA and other governmental agencies may be investigating our Abstral promotion practices. On December 16, 2015, we received a
subpoena issued by the U.S. Attorney’s Office in District of New Jersey requesting the production of a broad range of documents pertaining to our marketing and promotional practices for Abstral. We have been in contact with the U.S.
Attorney’s Office for the District of New Jersey and are cooperating in the production of the requested documents. We are unable to predict whether we could become subject to legal or administrative actions as a result of these matters, or the
impact of such matters. If we are found to be in violation of the False Claims Act or any other applicable state or and federal fraud and abuse laws, we may be subject to penalties, such as civil and criminal penalties, damages, fines, or an
administrative action of exclusion from government health care reimbursement programs. We can make no assurances as to the time or resources that will need to be devoted to these matters or their outcome, or the impact, if any, that these matters or
any resulting legal or administrative proceedings may have on our business or financial condition.
The federal False Claims Act
prohibits persons from knowingly filing, or causing to be filed, a false claim to, or the knowing use of false statements to obtain payment from, the federal government. Qui tam suits filed under the False Claims Act can be brought by any
individual on behalf of the government and such individuals, commonly known as “relators” or “whistleblowers,” may share in any amounts paid by the entity to the government in fines or settlement. The frequency of filing qui tam
actions has increased significantly in recent years, causing greater numbers of health care companies to have to defend such qui tam actions and pay substantial sums to settle such actions.
The federal Anti-Kickback Statute prohibits persons from knowingly and willfully soliciting, offering, receiving, or providing remuneration,
directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing or arranging for a good or service, for which payment may be made under a federal health care program such as the Medicare and Medicaid
programs. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal health care covered business, the statute has been violated.
The Anti-Kickback Statute is broad, and despite a series of narrow safe harbors, prohibits many arrangements and practices that are lawful in businesses outside of the health care industry. Penalties for violations of the federal Anti-Kickback
Statute include criminal penalties and civil and administrative sanctions such as fines, imprisonment and possible exclusion from Medicare, Medicaid and other federal health care programs. An alleged violation of the Anti- Kickback Statute may be
used as a predicate offense to establish liability pursuant to other federal laws and regulations such as the federal False Claims Act. Many states have also adopted laws similar to the federal Anti-Kickback Statute, some of which apply to the
referral of patients for health care items or services reimbursed by any source, not only Medicare and Medicaid programs.
S-11
The federal Patient Protection and Affordable Care Act includes provisions expanding the ability
of certain relators to bring actions that would have been dismissed under prior law. When an entity is determined to have violated the federal False Claims Act, it may be required to pay up to three times the actual damages sustained by the
government, plus civil penalties for each separate false claim. The Deficit Reduction Act of 2005 encouraged states to enact or modify their state false claims acts to be at least as effective as the federal False Claims Act by granting states a
portion of any federal Medicaid funds recovered through Medicaid-related actions. Most states have enacted state false claims laws, and many of those states included laws including qui tam provisions. The federal Patient Protection and Affordable
Care Act includes provisions known as the Physician Payments Sunshine Act, which requires manufacturers of drugs, biologics, devices and medical supplies covered under Medicare and Medicaid to record any transfers of value to physicians and teaching
hospitals and to report this data beginning in 2013 to the Centers for Medicare and Medicaid Services for subsequent public disclosures. Manufacturers must also disclose investment interests held by physicians and their family members. Failure to
submit the required information may result in civil monetary penalties of up to $1 million per year for knowing violations and may result in liability under other federal laws or regulations. Similar reporting requirements have also been enacted on
the state level in the U.S., and an increasing number of countries worldwide either have adopted or are considering similar laws requiring transparency of interactions with health care professionals. In addition, some states such as Massachusetts
and Vermont imposed an outright ban on certain gifts to physicians. These laws could affect our product promotional activities by limiting the kinds of interactions we could have with hospitals, physicians or other potential purchasers or users of
our system. Both the disclosure laws and gift bans also will impose administrative, cost and compliance burdens on us.
We face product liability
exposure and, if successful claims are brought against us, we may incur substantial liability if our insurance coverage for those claims is inadequate.
The commercial sale of our products after they are approved exposes us to possible product liability claims. This risk exists even if a
product is approved for commercial sale by the FDA and manufactured in facilities licensed and regulated by the FDA. Our products are designed to affect important bodily functions and processes. Any side effects, manufacturing defects, misuse or
abuse associated with our products could result in injury to a patient or even death. For example, because Abstral is designed to be self-administered by patients, it is possible that a patient could fail to follow instructions and as a result apply
a dose in a manner that results in injury. In addition, Abstral is an opioid pain reliever that contains fentanyl, which is regulated as a “controlled substance” under the Controlled Substances Act of 1970 (the “CSA”) and could
result in harm to patients relating to its potential for abuse. Product liability claims may be brought against us by consumers, health care providers, pharmaceutical companies or others selling or otherwise coming into contact with our products or
generic versions of our products. If we cannot successfully defend ourselves against product liability claims we could incur substantial liabilities. Because we have sold Abstral and Zuplenz, regardless of merit or eventual outcome, product
liability claims may result in:
| |
• |
|
impairment of our business reputation; |
| |
• |
|
costs of related litigation; |
| |
• |
|
distraction of management’s attention from our primary business; or |
| |
• |
|
substantial monetary awards to patients or other claimants. |
We have obtained product
liability insurance coverage for commercial product sales with a $10 million per occurrence and a $10 million annual aggregate coverage limit. We also carry excess product liability insurance coverage for commercial product sales with an additional
$5 million per occurrence and an additional $5 million annual aggregate coverage limit. Our insurance coverage may not be sufficient to cover all of our product liability related expenses or losses and may not cover us for any expenses or losses we
may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost, in sufficient amounts or upon adequate terms to protect us against losses due to
product liability. If we determine that it is prudent to increase our product liability coverage based on sales of our products, we may be unable to obtain this increased product liability insurance on commercially reasonable terms or at all. Large
judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects, including side effects that may be less severe than those of our products. A successful product liability claim or series of
claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and have a material adverse effect our business, results of operations, financial condition and prospects.
Our business involves the use of hazardous materials and we and our third-party manufacturers and suppliers must comply with environmental laws and
regulations, which can be expensive and restrict how we do business.
Our third-party manufacturers and suppliers activities
involve the controlled storage, use and disposal of hazardous materials. We and our manufacturers and suppliers are subject to laws and regulations governing the use, manufacture, storage, handling and
S-12
disposal of these hazardous materials even after we sell or otherwise dispose of the products. In some cases, these hazardous materials and various wastes resulting from their use were stored at
our contractors or manufacturers’ facilities pending use and disposal. We cannot completely eliminate the risk of contamination, which could cause injury to our employees and others, environmental damage resulting in costly clean-up and
liabilities under applicable laws and regulations governing the use, storage, handling and disposal of these materials and specified waste products. Although we expect that the safety procedures utilized by our third-party contractors and
manufacturers for handling and disposing of these materials will generally comply with the standards prescribed by these laws and regulations, we cannot guarantee that this will be the case or eliminate the risk of accidental contamination or injury
from these materials. In such an event, we may be held liable for any resulting damages and such liability could exceed our resources. We do not currently carry biological or hazardous waste insurance coverage and our property and casualty and
general liability insurance policies specifically exclude coverage for damages and fines arising from biological or hazardous waste exposure or contamination.
We will continue to be responsible for certain liabilities and obligations related to Abstral and Zuplenz, and if unknown liabilities were to arise it
could have a material adverse effect on us.
Under our respective asset purchase agreements with Sentynl and Midatech, our future
obligations under our former agreements with Orexo AB and MonoSol have been assumed by Sentynl and Midatech, respectively, except that we will continue to be responsible for chargebacks, rebates, patient assistance and certain other product
distribution channel liabilities related to Abstral and Zuplenz for a specified period of time post-closing. We also will be responsible for any pre-closing liabilities and obligations related to Abstral and Zuplenz, including unknown liabilities,
and have agreed in the respective asset purchase agreements to indemnify Sentynl and Midatech for any breach of our representations, warranties and covenants in the respective asset purchase agreements up to a certain agreed to amount. We cannot
quantify these responsibilities to Sentanyl and Midatech, but if substantial unknown liabilities were to arise, it could have a material adverse effect on our financial condition.
Risks Relating to Our Development Programs
Our
drug candidates may not receive regulatory approval or be successfully commercialized.
Before they can be marketed, our products
in development must be approved by the FDA or similar foreign governmental agencies. The process for obtaining FDA approval is both time-consuming and costly, with no certainty of a successful outcome. Before obtaining regulatory approval for the
sale of any drug candidate, we must conduct extensive preclinical tests and clinical trials to demonstrate the safety and efficacy in humans of our product candidates. Although our drug candidates have exhibited no serious adverse events
(“SAEs”) in the Phase 1 and 1/2 clinical trial, SAEs or other unexpected side effects may arising during further testing and development. A failure of any preclinical study or clinical trial can occur at any stage of testing. The results
of preclinical and initial clinical testing of these products may not necessarily indicate the results that will be obtained from later or more extensive testing. It also is possible to suffer significant setbacks in advanced clinical trials, even
after obtaining promising results in earlier trials.
Clinical trial designs that were discussed with the authorities prior to their commencement
may not result in the success of the trials or subsequently may not be considered sufficient for approval. Thus, our special protocol assessment with the FDA for our PRESENT trial does not ensure success of the trial or guarantee regulatory approval
of NeuVax for the treatment of breast cancer.
We reached agreement with the FDA regarding the special protocol assessment, or
SPA, for the design of our NeuVax Phase 3 PRESENT trial as an adjuvant in the treatment of patients with Node positive HER2 negative breast cancer in 2009. A SPA certifies the agreement with the FDA regarding the study endpoints, study design
and statistical assumptions of the clinical trial. The SPA is documented as part of the administrative record, and is binding on the FDA and may not be changed unless we fail to follow the agreed upon protocol, data supporting the test are found to
be false or incomplete, or the FDA determines that a substantial scientific issue essential to determining the safety or effectiveness of the drug was identified after the testing began. In June 2013, the FDA agreed to an amendment to the SPA to
account for the use of a companion diagnostic. Even with a SPA, approval of an NDA or a biological license application is not guaranteed because a final determination that an agreed upon protocol satisfies a specific objective, such as the
demonstration of efficacy, or supports an approval decision, will be based on a complete review of all the data submitted to the FDA. There is no assurance, therefore, that NeuVax Phase 3 PRESENT trial will be successful or that Neu Vax for the
treatment of patients with Node positive HER2 negative breast cancer will be approved by the FDA.
A number of different factors could prevent us
from obtaining regulatory approval or commercializing our product candidates on a timely basis, or at all.
We, the FDA or other
applicable regulatory authorities, an Independent Data Safety Monitoring Board or “IDSMB” governing our clinical trials, or an institutional review board, or “IRB,” which is an independent committee registered with and
S-13
overseen by the U.S. Department of Health and Human Services, or “HHS,” that functions to approve, monitor and review biomedical and behavioral research involving humans, may suspend
clinical trials of a drug candidate at any time for various reasons, including if we or it believe the subjects or patients participating in such trials are being exposed to unacceptable health risks. Among other reasons, adverse side effects of a
drug candidate on subjects or patients in a clinical trial could result in the FDA or other regulatory authorities suspending or terminating the trial and refusing to approve a particular drug candidate for any or all indications of use.
Clinical trials of a new drug candidate require the enrollment of a sufficient number of patients, including patients who are suffering from
the disease the drug candidate is intended to treat and who meet other eligibility criteria. Rates of patient enrollment are affected by many factors, and delays in patient enrollment can result in increased costs and longer development times than
we expect at present. Patients who are enrolled at the outset of this standard of care also may eventually choose for personal reasons not to participate in the study. We also compete for eligible patients with other breast cancer trials underway
from time to time, and we may experience delays in patient enrollment due to the dependency of other large trials underway in the same patient population.
Clinical trials also require the review and oversight of IRBs, which approve and continually review clinical investigations to protect the
rights and welfare of human subjects. An inability or delay in obtaining IRB approval could prevent or delay the initiation and completion of clinical trials, and the FDA may decide not to consider any data or information derived from a clinical
investigation not subject to initial and continuing IRB review and approval.
In addition, cancer vaccines are a relatively new form of
therapeutic treatment and a very limited number of such products have received regulatory approval. Therefore, the FDA or other regulatory authority may apply standards for approval of a new cancer vaccine that is different from past experience.
Numerous factors could affect the timing, cost or outcome of our drug development efforts, including the following:
| |
• |
|
difficulties or delays in enrolling patients in our Phase 1/2 clinical trials of GALE-301 (folate binding protein (FBP) vaccine), our Phase 2 clinical trial of GALE-401 (anagrelide controlled release) or other clinical
trials in conformity with required protocols or projected timeline or in our other NeuVax clinical trials; |
| |
• |
|
conditions imposed on us by the FDA, including the possibility that the FDA would require an additional Phase 3 trial of NeuVax, or comparable foreign authorities regarding the scope or design of our clinical trials;
|
| |
• |
|
difficulties or delays in arranging for third parties to conduct clinical trials of our product candidates; |
| |
• |
|
problems in engaging IRBs to oversee trials or problems in obtaining or maintaining IRB approval of studies; |
| |
• |
|
third-party contractors failing to comply with regulatory requirements or meet their contractual obligations to us in a timely manner; |
| |
• |
|
our drug candidates having very different chemical and pharmacological properties in humans than in laboratory testing and interacting with human biological systems in unforeseen, ineffective or harmful ways, and the
possibility that our previous Phase 2 trials will not be indicative of our drug candidates’ performance in larger patient populations; |
| |
• |
|
the need to suspend or terminate our clinical trials if the participants are being exposed to unacceptable health risks; |
| |
• |
|
insufficient or inadequate supply or quality of our drug candidates or other necessary materials necessary to conduct our clinical trials; |
| |
• |
|
disruption at our foreign clinical trial sites resulting from local social or political unrest or other geopolitical factors; |
| |
• |
|
effects of our drug candidates not being the desired effects or including undesirable side effects or the drug candidates having other unexpected characteristics; |
| |
• |
|
negative or inconclusive results from our clinical trials or the clinical trials of others for drug candidates similar to our own or inability to generate statistically significant data confirming the efficacy of the
product being tested; |
| |
• |
|
adverse results obtained by other companies developing similar drugs; |
S-14
| |
• |
|
modification of the drug during testing; and |
| |
• |
|
reallocation of our financial and other resources to other clinical programs. |
It is possible
that none of the product candidates that we develop will obtain the appropriate regulatory approvals necessary for us to begin selling them or that any regulatory approval to market a product may be subject to limitations on the indicated uses for
which we may market the product. The time required to obtain FDA and other approvals is unpredictable but often can take years following the commencement of clinical trials, depending upon the complexity of the drug candidate. Any analysis we
perform of data from clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. Any delay or failure in obtaining required approvals could have a material
adverse effect on our ability to generate revenue from the particular drug candidate.
In addition, the length of time to develop the
product candidates as well as any regulatory delays in the development and regulatory approval process could cause the patent exclusivity to be unavailable or greatly reduced for each product candidate. The lack of patent exclusivity could have a
material adverse effect on our ability to generate revenue from the particular drug candidate.
We are also subject to numerous foreign
regulatory requirements governing the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process includes all of the risks associated with the FDA approval
described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by the FDA does not assure approval by regulatory authorities outside of the U.S.
We are dependent upon contract manufacturers for clinical supplies of our product candidates, including our sole source of supply of a key component of
our Phase 3 PRESENT study of NeuVax.
We do not have the facilities or expertise to manufacture supplies of any of our product
candidates for clinical trials. Accordingly, we are dependent upon contract manufacturers for these supplies. There can be no assurance that we will be able to secure needed supply arrangements on reasonable terms, or at all. Our failure to secure
these arrangements as needed could have a materially adverse effect on our ability to complete the development of our product candidates or, if we obtain regulatory approval for our product candidates, to commercialize them.
Our current plans call for the manufacture of our compounds by contract manufacturers offering research grade, Good Laboratory Practices grade
and Good Manufacturing Practices grade materials for preclinical studies (e.g., toxicology studies) and for clinical use. Certain of our product candidates are complex molecules requiring many synthesis steps, which may lead to challenges with
purification and scale-up. These challenges could result in increased costs and delays in manufacturing.
NeuVax is administered in
combination with Leukine, a “GM-CSF” available in both liquid and lyopholyzed forms exclusively from Genzyme Corporation, or “Genzyme,” a subsidiary of Sanofi-Aventis. We will continue to be dependent on Genzyme for our supply of
Leukine in connection with the ongoing NeuVax and GALE-301 trials and the potential commercial manufacture of these programs. Any temporary interruptions or discontinuation of the availability of Leukine, or any determination by us to change the
GM-CSF used with NeuVax or GALE-301, may have a material adverse effect on our clinical trials and any commercialization of the assets.
We may not
be able to establish or maintain the third-party relationships that are necessary to develop or potentially commercialize some or all of our product candidates.
We expect to depend on collaborators, partners, licensees, clinical research organizations and other third parties to support our discovery
efforts, to formulate product candidates, to manufacture our product candidates, and to conduct clinical trials for some or all of our product candidates. We cannot guarantee that we will be able to successfully negotiate agreements for or maintain
relationships with collaborators, partners, licensees, clinical investigators, vendors and other third parties on favorable terms, if at all. Our ability to successfully negotiate such agreements will depend on, among other things, potential
partners’ evaluation of the superiority of our technology over competing technologies and the quality of the preclinical and clinical data that we have generated, and the perceived risks specific to developing our product candidates. If we are
unable to obtain or maintain these agreements, we may not be able to clinically develop, formulate, manufacture, obtain regulatory approvals for or commercialize our product candidates. Under certain license agreements that we have already entered
into, we have minimum dollar amounts per year that we are obligated to spend on the development of the technology we have licensed from our contract partners and other obligations to maintain certain licenses. If we fail to meet this requirement
under any of our licenses that contain such requirements or any other obligations under these licenses, we may be in breach of our obligations under such agreement, which may result in the loss of the technology licensed. We cannot necessarily
control the amount or timing of resources that our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will fulfill its obligations to
us under these arrangements in a timely fashion. We may not be able to readily terminate any such agreements with contract partners even if such contract partners do not fulfill its obligations to us.
S-15
In addition, we may receive notices from third parties from time to time alleging that our
technology or product candidates infringe upon the intellectual property rights of those third parties. Any assertion by third parties that our activities or product candidates infringe upon its intellectual property rights may adversely affect our
ability to secure strategic partners or licensees for our technology or product candidates or our ability to secure or maintain manufacturers for our compounds.
We are subject to competition and may not be able to compete successfully.
The biotechnology industry, including the cancer immunotherapy market, is intensely competitive and involves a high degree of risk. We compete
with other companies that have far greater experience and financial, research and technical resources than us. Potential competitors in the U.S. and worldwide are numerous and include pharmaceutical and biotechnology companies, educational
institutions and research foundations, many of which have substantially greater capital resources, marketing experience, research and development staffs and facilities than us. Some of our competitors may develop and commercialize products that
compete directly with those incorporating our technology, introduce products to market earlier than our products or on a more cost effective basis. In addition, our technology may be subject to competition from other technology or methods developed
using techniques other than those developed by traditional biotechnology methods. Our competitors compete with us in recruiting and retaining qualified scientific and management personnel as well as in acquiring technologies complementary to our
technology. Our collaborators or we will face competition with respect to product efficacy and safety, ease of use and adaptability to various modes of administration, acceptance by physicians, the timing and scope of regulatory approvals,
availability of resources, reimbursement coverage, price and patent position, including potentially dominant patent positions of others. An inability to successfully complete our product development could lead to us having limited prospects for
establishing market share or generating revenue from our technology.
For patients with early stage breast cancer, adjuvant therapy is
often given to prevent recurrence and increase the chance of long-term disease free survival. Adjuvant therapy for breast cancer can include chemotherapy, hormonal therapy, radiation therapy, or combinations thereof. In addition, the HER2 targeted
drug trastuzumab (Herceptin ) may be given to patients with tumors with high expression of HER2 (IHC 3+), in the adjuvant setting which may be useful in treating breast cancer.
There are a number of cancer vaccines in development for breast cancer, including but not limited to Lapuleucel-T (Dendreon), and AE-37
(Antigen Express). While these development candidates are aimed at a number of different targets, and AE-37 has published data in the HER2 breast cancer patient population, there is no guarantee that any of the these compounds will not in the future
be indicated for treatment of low to intermediate HER2 breast cancer patients and become directly competitive with NeuVax.
We are dependent on
technologies we license, and if we lose the right to license such technologies or we fail to license new technologies in the future, our ability to develop new products would be harmed.
We currently are dependent on licenses from third parties for technologies relating to our product candidates. Our current licenses impose,
and any future licenses we enter into are likely to impose, various development, funding, royalty, diligence, sublicensing, insurance and other obligations on us. If our license with respect to any of these technologies is terminated for any reason,
the development of the products contemplated by the licenses would be delayed, or suspended altogether, while we seek to license similar technology or develop new non-infringing technology. The costs of obtaining new licenses are high.
Risks associated with operating in foreign countries could materially adversely affect our product development.
We conduct our Phase 3 PRESENT study of NeuVax in countries outside of the U.S. Consequently, we are, and will continue to be, subject to
risks related to operating in foreign countries. Risks associated with conducting operations in foreign countries include:
| |
• |
|
differing regulatory requirements for drug approvals and regulation of approved drugs in foreign countries; |
| |
• |
|
unexpected changes in tariffs, trade barriers and regulatory requirements; economic weakness, including inflation, or political instability in particular foreign economies and markets; compliance with tax, employment,
immigration and labor laws for employees living or traveling abroad; foreign taxes, including withholding of payroll taxes; |
| |
• |
|
foreign currency fluctuations, which could result in increased operating expenses or reduced revenues, and other obligations incident to doing business or operating in another country; |
| |
• |
|
workforce uncertainty in countries where labor unrest is more common than in the U.S.; |
S-16
| |
• |
|
production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; and |
| |
• |
|
business interruptions resulting from geopolitical actions, including war and terrorism. |
In
addition, there may be political instability, including war, terrorism, riots, civil insurrection or social unrest, and natural or man-made disasters, including famine, flood, fire, earthquake, storm or disease, which could seriously harm the
progress of our clinical trials at sites in particular foreign countries or regions. For example, approximately 39 percent of our Phase 3 PRESENT trial sites and approximately 44% of patients who have been randomized in the trial are in Russia and
The Ukraine. The occupation of certain portions of Ukrainian territory by Russian-backed separatists and ongoing political and civil unrest there could disrupt activities at these trial sites. It is also possible that Russia could retaliate against
the imposition of sanctions by the U.S. and the European Union by banning or restricting business activities in Russia by U.S. companies, including the conduct of clinical trials, which could have a material, adverse effect on patient enrollment or
ongoing activities at our Phase 3 PRESENT sites in Russia.
Risks Relating to Our Financial Position and Capital Requirements
We may not be able to obtain sufficient financing, and may not be able to develop our product candidates.
We had cash and cash equivalents of approximately $34.8 million as of September 30, 2015. We believe that our existing cash and cash
equivalents together with the net proceeds of this offering should be sufficient to fund our operations for at least one year. This projection is based on our current planned operations, anticipated payments for defense costs and settlements or
judgments for the settlement and approval of the securities and derivative actions and opt-out cases, as well as the SEC investigation and other governmental investigations, and is subject to changes in our plans and uncertainties inherent in our
business, and we may need to seek to replenish our existing cash and cash equivalents sooner than we project. We also have funding available under our purchase agreement with Lincoln Park Capital Fund, LLC (subject to the lock-up restrictions
described below) and sales agreements with MLV & Co. and Maxim Group LLC described in the accompanying prospectus, but there is no guarantee that such funding will be available to us on favorable terms or will be sufficient to meet all of
our future funding needs. In connection with this offering, we have agreed that, without the prior written consent of Raymond James & Associates, Inc., for a period of 120 days following the date of this prospectus supplement, we will not
sell any shares of common stock pursuant to our purchase agreement with Lincoln Park Capital Fund, LLC. If we fail to obtain additional future funding when needed, we could be forced to scale back or terminate our operations, or to seek to merge
with or to be acquired by another company.
We expect to continue to incur significant research and development expenses, which may make it
difficult for us to attain profitability, and may lead to uncertainty about our ability to continue as a going concern.
Substantial funds were expended to develop our technologies and product candidates, and additional substantial funds will be required for
further preclinical testing and clinical trials of our product candidates, and to manufacture and market any products that are approved for commercial sale. Because the successful development of our products is uncertain, we are unable to precisely
estimate the actual funds we will require to develop and potentially commercialize them. In addition, we may not be able to generate enough revenue, even if we are able to commercialize any of our product candidates, to become profitable.
In the event that we are unable to achieve or sustain profitability or to secure additional financing, we may not be able to meet our
obligations as they come due, raising substantial doubts as to our ability to continue as a going concern. Any such inability to continue as a going concern may result in our common stock holders losing their entire investment. There is no guaranty
that we will become profitable or secure additional financing. Our financial statements contemplate that we will continue as a going concern and do not contain any adjustments that might result if we were unable to continue as a going concern.
Changes in our operating plans, our existing and anticipated working capital needs, the acceleration or modification of our expansion plans, increased expenses, potential acquisitions or other events will all affect our ability to continue as a
going concern. Future financing may be obtained through, and future development efforts may be paid for by, the issuance of debt or equity, which may have an adverse effect on our security holders or may otherwise adversely affect our business.
If we raise funds through the issuance of debt or equity, any debt securities or preferred stock issued will have rights, preferences and
privileges senior to those of holders of our common stock in the event of a liquidation. In such event, there is a possibility that once all senior claims are settled, there may be no assets remaining to pay out to the holders of common stock. In
addition, if we raise funds through the issuance of additional equity, whether through private placements or additional public offerings, such an issuance would dilute your ownership in us.
The terms of debt securities may also impose restrictions on our operations, which may include limiting our ability to incur additional
indebtedness, to pay dividends on or repurchase our capital stock, or to make certain acquisitions or investments. In addition, we may be subject to covenants requiring us to satisfy certain financial tests and ratios, and our ability to satisfy
such covenants may be affected by events outside of our control.
S-17
You may have difficulty evaluating our business, and our historical financial information may not be
representative of our future results.
We recently sold our Abstral and Zuplenz products and related assets and discontinued our
commercial operations as of December 31, 2015 in order to focus our resources on our pipeline of product candidates. As a result, we will have no recurring revenues unless and until we are able to obtain marketing approval of NeuVax or one or
more of our other product candidates and our historical financial information may not be representative of our future results.
We may be unable to
comply with our reporting and other requirements under federal securities laws.
As a publicly traded company, we are subject to
the reporting requirements of the Securities Exchange Act of 1934, as amended, or the “Exchange Act,” and the Sarbanes-Oxley Act of 2002, or the “Sarbanes-Oxley Act.” In addition, the Exchange Act requires that we file annual,
quarterly and current reports. Our failure to prepare and disclose this information in a timely manner could subject us to penalties under federal securities laws, expose us to lawsuits and restrict our ability to access financing. The
Sarbanes-Oxley Act requires that we, among other things, establish and maintain effective internal controls and procedures for financial reporting. From time to time we evaluate our existing internal controls in light of the standards adopted by the
Public Company Accounting Oversight Board. It is possible that we or our independent registered public accounting firm may identify significant deficiencies or material weaknesses in our internal control over financial reporting in the future. Any
failure or difficulties in implementing and maintaining these controls could cause us to fail to meet the periodic reporting obligations or result in material misstatements in our financial statements.
Section 404 of the Sarbanes-Oxley Act requires annual management and independent auditor assessments of the effectiveness of our internal
control over financial reporting. Our failure to satisfy the requirements of Section 404 on a timely basis could result in the loss of investor confidence in the reliability of our financial statements, which in turn could have a material
adverse effect on our business and our common stock.
Risks Related to Our Intellectual Property
We may not be able to obtain and enforce patent rights or other intellectual property rights that cover our product candidates and that are of sufficient
breadth to prevent third parties from competing against us.
Our success with respect to our product candidates will depend in
part on our ability to obtain and maintain patent protection in the U.S. and abroad, to preserve our trade secrets, and to prevent third parties from infringing upon our proprietary rights. Our patents and patent applications, however, may not be
sufficient to provide protection for NeuVax or our other products and product candidates against commercial competition.
The active
peptide found in NeuVax, the E75 peptide, has been known and studied for many years. We have one issued U.S. patent, US 6,514,942, covering the composition of matter of the E75 peptide, which expired in mid-2015, prior to any potential
commercialization of NeuVax. We do not have and will not be able to obtain any composition of matter patent protection for E75, the active peptide in NeuVax. We also have a license from The Henry M. Jackson Foundation to an issued U.S. European,
Japanese and Australian method of use patents, which expire in 2028, that are directed to a method of inducing immunity against breast cancer recurrence by administering a composition comprising the E75 peptide to patients who have both an
immunohistochemistry (IHC) rating of 1+ or 2+ for HER2/neu protein expression and a fluorescence in situ hybridization (FISH) rating of less than about 2.0 for HER2/neu gene expression. The license further includes an issued U.S. method of use
patent, and pending applications in a number of foreign jurisdictions, which expires in 2028, that are directed to a method of inducing immunity against recurrence of any HER2/neu expressing tumors by administering the E75 peptide to patients with
tumors having a FISH rating of less than about 2.0 for HER2/neu gene expression. Also included in the license is a method of use patent, which expires in 2026, that is directed to the use of NeuVax in combination with Herceptin® to treat any HER2/neu expressing cancer. Thus, our method of use patent may not prevent competitors from seeking to develop and market NeuVax for use in cancer patients who do not meet these
criteria. If any such alternative uses were approved, this could lead to off-label use and price erosion for our NeuVax product. We may seek FDA approval for use of NeuVax to treat cancer patients who fall outside the claimed IHC and FISH ranges and
for other cancers as well. Although we are pursuing additional patent protection for NeuVax through pending patent applications, we may not be able to obtain additional patent protection that would provide us with a significant commercial advantage.
Anagrelide hydrochloride, the sole active pharmaceutical ingredient, or “API,” in GALE-401, has been approved for many years
and, thus, it is not possible to obtain composition of matter patents that cover anagrelide hydrochloride. As a result, competitors
S-18
who obtain the requisite regulatory approval can offer products with the same API as GALE-401, so long as the competitors do not infringe any formulation patents that we may have or may obtain or
license, if any. The only patent protection that we have or are likely to obtain covering GALE-401 are patents relating to very specific formulations, methods using these formulations, and methods of manufacturing and packaging. We have an issued
U.S. Patent, which expires in 2020, covering methods of anagrelide to reduce platelet count in patients subject to veno-occlusive events. We have granted patents in the U.S., United Kingdom and Japan, which expire in 2029, covering controlled
release formulations of anagrelide and methods of use. We are also prosecuting pending patent applications in other territories including but not limited to the U.S. Europe, and Japan, which may not issue prior to any potential commercialization of
GALE-401. We may seek FDA approval for use of GALE-401 to treat patients with myeloproliferative neoplasms that include several hermatological disorders. Although we are pursuing additional patent protection for GALE-401 through pending patent
applications, we may not be able to obtain additional patent protection that would provide us with a significant commercial advantage.
The active peptides found in GALE-301 and GALE-302 are derived from Folate Binding Protein. One of the active peptides, E39, has been known
and studied for many years. The other active peptide, GALE-302, is a derivative of E39. We have a license from The Henry M. Jackson Foundation to issued and granted patents in the U.S., Europe, Canada, and Japan, covering composition of matter for
the E39 derivative peptides, including GALE-302, alone and in combination with E39, as well as the use of these compositions for the treatment of cancer. These patents are expected to expire in 2022, prior to any potential commercialization of
GALE-301. We do not have and will not be able to obtain any composition of matter patent protection for the E39 peptide in any territory. The license we have from The Henry M. Jackson Foundation grants us the right to develop and market GALE-301 for
any use, including methods of treating cancer, our patents may not prevent competitors from seeking to develop and market the E39 peptide alone. If any such alternative uses of compositions containing the E39 peptide were approved, this could lead
to off-label use and price erosion for GALE-301. We may seek FDA approval for use of GALE-301, alone or in combination with GALE-302, to treat cancer patients with ovarian and endometrial cancers and for other cancers, as well. Although we are
pursuing additional patent protection for GALE-301 and the combination of GALE-301 and GALE-302 through pending patent applications, we may not be able to obtain additional patent protection that would provide us with a significant commercial
advantage.
Our ability to obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions.
Accordingly, rights under any patents we have or may obtain or license may not provide us with sufficient protection for our commercial product and product candidates to afford a commercial advantage against competitive products or processes,
including those from branded and generic pharmaceutical companies. In addition, we cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us. Nor can we guarantee that the claims of these
patents will be held valid or enforceable by the courts or will provide us with any significant protection against competitive products or otherwise be commercially valuable to us.
Changes in either the patent laws or in the interpretations of patent laws in the U.S. or abroad may diminish the value of our intellectual
property. In addition, on September 16, 2011, the Leahy-Smith America Invents Act, or the Leahy-Smith Act, was signed into law. The Leahy-Smith Act includes a number of significant changes to the U.S. patent law. These include provisions that
affect the way patent applications will be prosecuted and may also affect patent litigation. The U.S. Patent Office is currently developing regulations and procedures to govern administration of the Leahy-Smith Act, and many substantive changes to
patent law associated with the Leahy-Smith Act have not yet become effective. Accordingly, it is not clear what, if any, impact the Leahy-Smith Act will have on the operation of our business. However, the Leahy-Smith Act, in particular the
first-to-file provision and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement of or defense of our issued patents, all of which could have a material adverse
effect on our business and financial condition. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents.
While we intend to take actions reasonably necessary to enforce our patent rights, we may not be able to detect infringement of our own or
in-licensed patents, which may be especially difficult for methods of manufacturing or formulation products, and we depend, in part, on our licensors and collaborators to protect a substantial portion of our proprietary rights. In addition, third
parties may challenge our in-licensed patents and any of our own patents that we may obtain, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. Litigation or other proceedings to enforce or
defend intellectual property rights is very complex, expensive, and may divert our management’s attention from our core business and may result in unfavorable results that could adversely affect our ability to prevent third parties from
competing with us.
If another party has reason to assert a substantial new question of patentability against any of our claims in our own
and in-licensed patents, the third party can request that the patent claims be reexamined, which may result in a loss of scope of some claims or a loss of the entire patent. In addition to potential infringement suits and, interference and
reexamination proceedings, we may become a party to patent opposition proceedings where either the patentability of the inventions subject of our patents are challenged, or we are challenging the patents of others. The costs of these proceedings
could be substantial, and it is possible that such efforts would be unsuccessful.
S-19
As the medical device, biotechnology and pharmaceutical industries expand and more patents are
issued, the risk increases that others may assert our commercial product and/or product candidates infringe their patent rights. If a third-party’s patents were found to cover our commercial product and product candidates, proprietary
technologies or its uses, we or our collaborators could be enjoined by a court and required to pay damages and could be unable to continue to commercialize our products or use our proprietary technologies unless we or it obtained a license to the
patent. A license may not be available to us or our collaborators on acceptable terms, if at all. In addition, during litigation, the patent holder could obtain a preliminary injunction or other equitable relief, which could prohibit us from making,
using or selling our commercial product and product candidates pending a trial on the merits, which could be years away.
Proprietary
trade secrets and unpatented know-how are also very important to our business. Although we have taken steps to protect our trade secrets and unpatented know-how, by entering into confidentiality agreements with third parties, and proprietary
information and invention agreements with certain employees, consultants and advisors, third parties may still obtain this information or we may be unable to protect our rights. We also have limited control over the protection of trade secrets used
by our licensors, collaborators and suppliers. There can be no assurance that binding agreements will not be breached, that we would have adequate remedies for any breach, or that our trade secrets and unpatented know-how will not otherwise become
known or be independently discovered by our competitors. If trade secrets are independently discovered, we would not be able to prevent their use. Enforcing a claim that a third party illegally obtained and is using our trade secrets or unpatented
know-how is expensive and time consuming, and the outcome is unpredictable.
We may be subject to claims that our employees, consultants
or independent contractors have wrongfully used or disclosed to us alleged trade secrets of their other clients or former employers. As is common in the biotechnology and pharmaceutical industry, certain of our employees were formerly employed by
other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Moreover, we engage the services of consultants to assist us in the development of our commercial product and product candidates, many of whom were
previously employed at or may have previously been or are currently providing consulting services to, other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these
employees and consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers or their former or current customers. Litigation may be necessary to defend against these
types of claims. Even if we are successful in defending against any such claims, any such litigation would likely be protracted, expensive, a distraction to our management team, not viewed favorably by investors and other third parties, and may
potentially result in an unfavorable outcome.
Our product candidates may face competition sooner than expected after the expiration of our
composition of matter patent protection for such products.
Our composition of matter patents for many our product candidates have
expired will expire prior to any product approval. We intend to seek data exclusivity or market exclusivity for our NeuVax, GALE-301 and GALE-302 product candidates provided under the Federal Food, Drug and Cosmetic Act, or FDCA, and similar laws in
other countries. We believe that these product candidates will qualify for 12 years of data exclusivity under the Biologics Price Competition and Innovation Act of 2009, or BPCIA, which was enacted as part of the Patient Protection and Affordable
Care Act and the Health Care and Education Affordability Reconciliation Act of 2010 (collectively, the Affordable Care Act or ACA) enacted in March 2010. Under the BPCIA, an application for a biosimilar product or biologics license application (BLA)
cannot be submitted to the FDA until four years, or if approved by the FDA, until 12 years, after the original brand product identified as the reference product is approved under a BLA. The BPCIA provides an abbreviated pathway for the approval of
biosimilar and interchangeable biological products. The new abbreviated regulatory pathway establishes legal authority for the FDA to review and approve biosimilar biologics, including the possible designation of a biosimilar as
“interchangeable” based on its similarity to an existing brand product. The new law is complex and is only beginning to be interpreted and implemented by the FDA. While it is uncertain when any such processes may be fully adopted by the
FDA, any such processes could have a material adverse effect on the future commercial prospects for our biological product candidates. There is also a risk that the U.S. Congress could amend the BPCIA to shorten this exclusivity period as proposed
by President Obama, potentially creating the opportunity for biosimilar competition sooner than anticipated after the expiration of our patent protection. Moreover, the extent to which a biosimilar, once approved, will be substituted for any
reference product in a way that is similar to traditional generic substitution for non-biological products is not yet clear, and will depend on a number of marketplace and regulatory factors that are still developing.
If our product candidates are not considered biologics that would qualify for exclusivity under the BPCIA, they may be eligible for market
exclusivity as drugs under the FDCA. The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has
not previously approved any other new drug containing the same active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug
application, or ANDA, or a 505(b)(2) NDA, submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be
submitted after four years if it
S-20
contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an existing NDA if new
clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by FDA to be essential to the approval of the application, for example, for new indications, dosages, or strengths of an
existing drug. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for drugs containing the original active agent.
Even if, as we expect, NeuVax, GALE-301 and GALE-302 are considered to be reference products eligible for 12 years of exclusivity under the
BPCIA or five years of exclusivity under the FDCA, another company could market competing products if the FDA approves a full BLA or full NDA for such product containing the sponsor’s own preclinical data and data from adequate and
well-controlled clinical trials to demonstrate the safety, purity and potency of the products.
In some countries outside of the U.S.,
peptide vaccines, such as NeuVax and GALE-301 and GALE-302, are regulated as chemical drugs rather than as biologics and may or may not be eligible for non-patent exclusivity.
Risks Relating to Ownership of Our Common Stock
The market price and trading volume of our common stock may be volatile.
The market price of our common stock has exhibited substantial volatility recently. Between January 1, 2015 and January 6, 2016, the
sale price of our common stock as reported on The NASDAQ Capital Market ranged from a low of $1.29 to a high of $2.24. The market price of our common stock could continue to fluctuate significantly for many reasons, including the following factors:
| |
• |
|
reports of the results of our clinical trials regarding the safety or efficacy of our product candidates and surrogate markers; |
| |
• |
|
announcements of regulatory developments or technological innovations by us or our competitors; |
| |
• |
|
announcements of business or strategic transactions such as our recent sales of our marketed products and discontinuation of our commercial operations, or our success in doing so; |
| |
• |
|
announcements of legal or regulatory actions against us or any adverse outcome of any such actions; |
| |
• |
|
changes in our relationship with our licensors, licensees and other strategic partners; |
| |
• |
|
our quarterly operating results; |
| |
• |
|
developments in patent or other technology ownership rights; |
| |
• |
|
public concern regarding the safety of our product candidates; |
| |
• |
|
additional funds may not be available on terms that are favorable to us and, in the case of equity financings, may result in dilution to our stockholders; |
| |
• |
|
government regulation of drug pricing; and |
| |
• |
|
general changes in the economy, the financial markets or the pharmaceutical or biotechnology industries. |
Factors beyond our control may also have an impact on the price of our stock. For example, to the extent that other companies within our
industry experience declines in their stock prices, our stock price may decline as well.
We are, and in the future may be, subject to legal or
administrative actions that could adversely affect our financial condition and our business.
On December 3, 2015, we
agreed in principle to resolve and settle the consolidated shareholder derivative action, In re Galena Biopharma, Inc. Derivative Litigation, Civil Action No. 3:14-cv-00382-SI, currently pending in the United States District Court for
the District of Oregon against us and certain of our current and former officers and directors. The settlement will not become effective until approved by the Court. The settlement includes a payment of $15 million in cash by our insurance carriers,
which we will use to fund a portion of the class action settlement, and cancellation of 1,200,000 outstanding director stock options.
S-21
The settlement also will require that we adopt and implement certain corporate governance measures and will provide that the plaintiffs’ counsel may apply to the court for an award of
attorneys’ fees and expenses up to $5 million. Any fees and expenses awarded by the court to the plaintiffs’ counsel will be paid by our insurance carriers. The settlement will not include any admission of wrongdoing or liability on the
part of us or the individual defendants and will include a full release of us and the individual defendants in connection with the allegations made in the consolidated federal derivative actions and state court derivative actions.
On December 3, 2015, we also agreed in principal to resolve and settle the securities putative class action lawsuit, In re
Galena Biopharma, Inc. Securities Litigation, Civil Action No. 3:14-cv-00367-SI, pending against us, certain of our current and former officers and directors and other defendants in the United States District Court for the District of
Oregon. The agreement, which is subject to shareholder notice and Court approval, provides for a settlement payment of $20 million to the class and the dismissal of all claims against us and the other defendants in connection with the consolidated
federal securities class actions. Of the $20 million settlement payment to the class, $16.7 million will be paid by our insurance carriers and $3.3 million will be paid by us through a combination of $2.3 million in cash and $1 million in shares of
our common stock. We will be responsible for defense costs and any settlements or judgments incurred for any related opt-out lawsuits.
We are aware that the SEC is investigating certain matters relating to the use of certain outside investor-relations professionals by us and
other public companies. We have been in contact with the SEC staff through our counsel and are cooperating with the investigation.
Litigation is inherently uncertain. We have incurred and may continue to incur substantial unreimbursed legal fees and other expenses in
connection with these or other legal and regulatory proceedings that may not qualify for coverage under, or may exceed the limits of, our applicable directors and officers liability insurance policies and could have a material adverse effect on our
financial condition, liquidity, and results of operations. These matters also may distract the time and attention of our officers and directors or divert our other resources away from our ongoing commercial and development programs. An unfavorable
outcome in any of these matters could damage our business and reputation or result in additional claims or proceedings against us.
The Delaware
forum provision of our amended and restated by-laws will not be given effect.
On August 6, 2013, our board of directors
purported to adopt an amendment to our Amended and Restated By-Laws to add a new Section 6.15 to provide that, unless we consent in writing to the selection of an alternative forum, the Court of Chancery of the State of Delaware will be the
sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders. Under the Delaware General Corporation Law, or DGCL, if the board of directors of a Delaware corporation such as our company is intended
to have the power to adopt and amend the bylaws, that power must be set forth in the corporation’s certificate of incorporation. Our amended and restated certificate of incorporation does not provide for the power of our board to adopt and
amend the bylaws, and we have determined that the Delaware forum bylaw is inconsistent with the DGCL and will not be given effect.
Future sales of
substantial amounts of our common stock, or the possibility that such sales could occur, could adversely affect the market price of our common stock.
Future sales in the public market of shares of our common stock, including shares referred to in the foregoing risk factors or shares issued
upon exercise of our outstanding stock options, or the perception by the market that these sales could occur, could lower the market price of our common stock or make it difficult for us to raise additional capital.
As of September 30, 2015, we had reserved for issuance 11,188,443 shares of our common stock issuable upon the exercise of outstanding
stock options at a weighted-average exercise price of $2.83 per share and 22,308,475 shares of our common stock issuable upon the exercise of outstanding warrants at a weighted-average exercise price of $2.10 per share. Upon exercise of these
options and warrants, the underlying shares may be resold into the public market. In the case of outstanding options and warrants that have exercise prices that are below the market price of our common stock from time to time, our stockholders would
experience dilution upon the exercise of these options.
Our outstanding warrants may result in dilution to our stockholders.
Our outstanding March 2011 and April 2011 warrants to purchase a total of 791,398 shares of common stock as of September 30, 2015 at a
current exercise price of $0.65 per share contain so-called full-ratchet anti-dilution provisions. Our outstanding March 2010 and December 2012 warrants to purchase 25,000 shares and 3,031,311 shares, respectively, of common stock as of
September 30, 2015 at exercise prices of $2.08 and $1.83, respectively, per share contain so-called weighted-average anti-dilution provisions. These anti-dilution provisions may be triggered by the issuance of the shares being offered hereby or
upon any future issuance by us of shares of our common stock or common stock equivalents at a price per share below the then-exercise price of the warrants, subject to some exceptions.
S-22
To the extent that these anti-dilution provisions are triggered in the future, we would be
required to reduce the exercise price of all of the warrants on either a full-ratchet or weighted-average basis, which would have a dilutive effect on our stockholders.
We may issue preferred stock in the future, and the terms of the preferred stock may reduce the value of our common stock.
We are authorized to issue up to 5,000,000 shares of preferred stock in one or more series. Our board of directors may determine the terms of
future preferred stock offerings without further action by our stockholders. If we issue preferred stock, it could affect stockholder rights or reduce the market value of our outstanding common stock. In particular, specific rights granted to future
holders of preferred stock may include voting rights, preferences as to dividends and liquidation, conversion and redemption rights, sinking fund provisions, and restrictions on our ability to merge with or sell our assets to a third party.
Anti-takeover provisions of our amended and restated certificate of incorporation and amended and restated bylaws and provisions of Delaware law could
delay or prevent a change of control that our stockholders may favor.
Anti-takeover provisions of our amended and restated
certificate of incorporation and amended and restated bylaws may discourage, delay or prevent a merger or other change of control that stockholders may consider favorable or may impede the ability of the holders of our common stock to change our
management. These provisions of our amended and restated certificate of incorporation and amended and restated bylaws, among other things:
| |
• |
|
divide our board of directors into three classes, with members of each class to be elected for staggered three-year terms; |
| |
• |
|
limit the right of security holders to remove directors; |
| |
• |
|
prohibit stockholders from acting by written consent; |
| |
• |
|
regulate how stockholders may present proposals or nominate directors for election at annual meetings of stockholders; and |
| |
• |
|
authorize our board of directors to issue preferred stock in one or more series, without stockholder approval. |
In addition, Section 203 of the Delaware General Corporation Law provides that, subject to limited exceptions, persons that acquire, or
are affiliated with a person that acquires, more than 15% of the outstanding voting stock of a Delaware corporation such as our company shall not engage in any business combination with that corporation, including by merger, consolidation or
acquisitions of additional shares for a three-year period following the date on which that person or its affiliate crosses the 15% stock ownership threshold. Section 203 could operate to delay or prevent a change of control of our company.
We have never declared or paid cash dividends on our capital stock and we do not anticipate paying cash dividends in the foreseeable future.
Our business requires significant funding. We currently plan to invest all available funds and future earnings in the development
and growth of our business and do not anticipate paying any cash dividends on our common stock in the foreseeable future, and are prohibited by the terms of our outstanding indebtedness from paying dividends on any common stock, except with the
prior consent of our lenders. As a result, capital appreciation, if any, of our common stock will be our stockholders’ sole source of potential gain for the foreseeable future.
The terms of our outstanding indebtedness may inhibit potential acquirers.
We are prohibited by the terms of our outstanding indebtedness from disposing of any of our business or property, except with the consent of
our lenders or if we were to prepay the outstanding indebtedness and related fees in accordance with the loan security agreement. Our outstanding indebtedness may inhibit potential acquirers or other interested parties from seeking to acquire all or
a part of our business or assets, and there is no assurance that our lenders would consent to any proposed future transaction that might be beneficial to our stockholders.
S-23
Risks Relating to this Offering
Management will have broad discretion as to the use of the net proceeds of this offering.
We currently intend to use the net proceeds from this offering to fund our ongoing Phase 3 PRESENT study and other clinical trials of our
product candidates, and to augment our working capital and for general corporate purposes. We have not reserved or allocated amounts for any specific purposes, however, and we cannot specify with certainty how we will use any net proceeds.
Accordingly, our management will have considerable discretion in the application of the net proceeds and you will not have the opportunity, as part of your investment decision, to assess whether the proceeds are being used appropriately. The net
proceeds of this offering may be used for corporate purposes that do not benefit our company or increase our market value. Until the net proceeds are used, they may be placed in investments that may not produce income or that may lose their value.
Investors will experience immediate and substantial dilution.
You will suffer immediate and substantial dilution. See the “Dilution” section in this prospectus supplement for more information
about the dilution you will incur in this offering.
You may not be able to resell your warrants.
There is no established trading market for the warrants being offered in this offering, and we do not expect such a market to develop. In
addition, we do not intend to apply for listing of the warrants on any securities exchange or other nationally recognized trading system, and you may not be able to resell your warrants. If your warrants cannot be resold, you will have to depend
upon any appreciation in the value of our common stock over the exercise price of the warrants in order to realize a return on your investment in the warrants.
Investors will have no rights as a common stockholder with respect to their warrants until they exercise their warrants and acquire our common stock.
Until you acquire shares of our common stock upon exercise of your warrants, you will have no rights with respect to the shares
of our common stock underlying such warrants. Upon exercise of your warrants, you will be entitled to exercise the rights of a common stockholder only as to matters for which the record date occurs after the exercise date.
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus supplement, the accompanying prospectus and the other documents we have filed with the SEC that are incorporated herein by
reference contain forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 that involve risks and uncertainties, as well as assumptions that, if they never materialize or prove incorrect, could cause our
results to differ materially from those expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be deemed forward-looking statements, including any projections of
financing needs, revenue, expenses, earnings or losses from operations, or other financial items, any statements of the plans, strategies and objectives of management for future operations, any statements concerning product research, development and
commercialization plans and timelines, any statements regarding safety and efficacy of product candidates, any statements of expectation or belief and any statements of assumptions underlying any of the foregoing. In addition, forward-looking
statements may contain the words “believe,” “anticipate,” “expect,” “estimate,” “intend,” “plan,” “project,” “will be,” “will continue,” “will
result,” “seek,” “could,” “may,” “might,” or any variations of such words or other words with similar meanings. All forward-looking statements attributable to us or to persons acting on our behalf are
expressly qualified in their entirety by the cautionary statements and risk factors set forth in the “Risk Factors” section and elsewhere in this prospectus supplement, in the accompanying prospectus and in our Annual Report on Form 10-K
for the year ended December 31, 2014 and our subsequent Quarterly Reports on Form 10-Q.
Given these uncertainties, you should not
place undue reliance on these forward-looking statements. You should read this prospectus supplement, the accompanying prospectus and the documents incorporated by reference in this prospectus supplement and the accompanying prospectus with the
understanding that our actual future results may be materially different from what we expect. Except as required by law, we do not undertake any obligation to update or revise any forward-looking statements contained in this prospectus supplement,
the accompanying prospectus or such other documents, whether as a result of new information, future events or otherwise.
S-24
USE OF PROCEEDS
We estimate that the net proceeds from this offering, after deducting underwriting discounts and commissions and estimated offering expenses
payable by us, will be approximately $20.145 million (or approximately $23.2 million if the underwriters’ over-allotment option is exercised in full).
Except as otherwise described in any free writing prospectus that we may authorize to be furnished to you, we currently intend to use the net
proceeds from this offering to fund our ongoing Phase 3 PRESENT study and other clinical trials of our product candidates, and to augment our working capital for general corporate purposes. General corporate purposes may include, but are not limited
to, payments relating to the settlement of our shareholder lawsuits and discontinuation of our commercial operations, repayment of existing or future indebtedness, financing of capital expenditures and potential future acquisitions and strategic
investments. Pending application of the net proceeds, we expect to invest the net proceeds in investment-grade, augment our working capital and for general corporate purposes, interest-bearing securities.
We had outstanding as of September 30, 2015 $5.7 million principal amount of indebtedness incurred in May 2013 under our loan and security
agreement with Oxford Finance LLC, as collateral agent, the proceeds of which were used to replenish our working capital following our acquisition on March 18, 2013 of U.S. rights in Abstral for which we paid the seller $10 million up front.
Our monthly payments on the outstanding indebtedness under the loan and security agreement consist of amortization of principal together with interest at a fixed annual rate of 8.45% until maturity in November 2016.
We have not determined the amounts we plan to spend on any of the areas listed above or the timing of these expenditures. As a result, our
management will have broad discretion to allocate the net proceeds from this offering. Pending application of the net proceeds as described above, we expect to invest the net proceeds in short-term, interest-bearing, investment-grade securities.
DIVIDEND POLICY
Our business requires significant funding. We currently plan to invest all available funds and any future earnings in our business and do not
anticipate paying any cash dividends on our common stock in the foreseeable future. We currently are prohibited by the terms of our outstanding indebtedness from paying dividends on our common stock, except with the prior consent of our lenders.
DILUTION
Our net tangible book value (deficit) as of September 30, 2015 was approximately ($9.5 million), or ($0.06) per share of common stock.
Net tangible book value (deficit) per share is calculated by subtracting our total liabilities from our total tangible assets, which is total assets less intangible assets, and dividing this amount by the number of shares of common stock
outstanding. After giving effect to the sale of 19,772,727 units in this offering (excluding 11,863,636 shares of common stock issuable upon exercise of the warrants being offered in this offering) at the public offering price of $1.10 per unit and
after deducting underwriting discounts and commissions and estimated offering expenses payable by us, we would have had a net tangible book value as of September 30, 2015 of approximately $10.6 million, or $0.06 per share of common stock.
This represents an immediate increase in net tangible book value of $0.12 per share to our existing stockholders and an immediate dilution in net tangible book value of $1.04 per share to investors in this offering. The following table illustrates
this dilution:
|
|
|
|
|
|
|
|
|
| Public offering price per unit |
|
|
|
|
|
$ |
1.10 |
|
| Net tangible book value (deficit) per share as of September 30, 2015 |
|
($ |
0.06 |
) |
|
|
|
|
| Increase per share attributable to this offering |
|
$ |
0.12 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| As adjusted net tangible book per share after this offering |
|
|
|
|
|
$ |
0.06 |
|
|
|
|
|
|
|
|
|
|
| Net dilution per share to investors in this offering |
|
|
|
|
|
$ |
1.04 |
|
|
|
|
|
|
|
|
|
|
If the underwriters’ over-allotment option is exercised in full, our as adjusted net tangible book value
at September 30, 2015 would have been approximately $13.7 million, or approximately $0.07 per share of common stock, and the dilution to investors in this offering would have been approximately $1.03 per share.
The number of shares of common stock shown above to be outstanding after this offering is based on 161,900,446 shares outstanding as of
September 30, 2015 and excludes as of such date:
| |
• |
|
675,000 shares held in treasury; |
S-25
| |
• |
|
11,188,443 shares of our common stock subject to outstanding options having a weighted-average exercise price of $2.83 per share; |
| |
• |
|
10,250,759 shares of our common stock reserved for issuance in connection with future awards under our 2007 Stock Incentive Plan; |
| |
• |
|
528,131 shares of our common stock reserved for future sale under our employee stock purchase plan; |
| |
• |
|
22,308,475 shares of our common stock subject to outstanding warrants having a weighted-average exercise price of $2.10 per share; and |
| |
• |
|
11,863,636 shares of common stock issuable upon exercise of warrants to be issued in this offering. |
The shares of common stock issuable upon the exercise of our outstanding warrants and the exercise price of the warrants are subject to an
adjustment in certain circumstances. Our outstanding March 2011 and April 2011 warrants to purchase a total of 791,398 shares of common stock as of September 30, 2015 at a current exercise price of $0.65 per share contain so-called full-ratchet
anti-dilution provisions. Our outstanding March 2010 and December 2012 warrants to purchase 25,000 shares and 3,031,311 shares, respectively, of common stock as of September 30, 2015 at exercise prices of $2.08 and $1.83, respectively, per
share contain so-called weighted average anti-dilution provisions. These anti-dilution provisions would be triggered upon any issuance by us of shares of our common stock or common stock equivalents at a price per share below the then-exercise price
of warrants, subject to some exceptions. Upon consummation of the offering, we anticipate that the exercise price of our outstanding December 2012 warrants to purchase a total of 3,031,311 shares of common stock as of September 30, 2015 will be
adjusted downward from $1.83 to $1.75 per share that the exercise price of our outstanding March 2010 warrants to purchase a total of 25,000 shares of common stock as of September 30, 2015 will be adjusted downward from $2.02 to $1.92 per
share.
To the extent our outstanding options and warrants are exercised, you may experience further dilution. The above illustration of
dilution per share to investors participating in this offering assumes no exercise of outstanding options or outstanding warrants to purchase shares of our common stock. The illustration also assumes no further issuance of shares of our common stock
in payment of contingent consideration to holders of our outstanding contingent value rights or others. The exercise of outstanding options and warrants having an exercise price less than the offering price of the units in this offering, or our
payment to our contingent value rights holders or others of common shares valued at less than the offering price of units in this offering, would further increase dilution to investors in this offering.
S-26
CAPITALIZATION
The following table sets forth our cash and cash-equivalents and our capitalization as of September 30, 2015 as follows:
| |
• |
|
On an actual basis; and |
| |
• |
|
On an as-adjusted basis to give effect to our issuance and sale of 19,772,727 units in this offering at the public offering price of $1.10 per unit, after deducting the estimated underwriting discounts and commissions
and estimated offering expenses payable by us, assuming no exercise of the underwriters’ over-allotment option and excluding the proceeds, if any, from the exercise of warrants issued pursuant to this offering. |
You should read the information in the following table in conjunction with our consolidated financial statements and the related notes and the
section entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” contained in our Quarterly Report on Form 10-Q for the quarter ended September 30, 2015 filed with the SEC and incorporated
by reference in this prospectus supplement and the accompanying prospectus.
|
|
|
|
|
|
|
|
|
| |
|
As of September 30, 2015 |
|
| (In thousands, except share and per share information) |
|
Actual |
|
|
As adjusted for this
offering |
|
| Cash and cash equivalents |
|
$ |
34,812 |
|
|
$ |
54,947 |
|
|
|
|
|
|
|
|
|
|
|
|
|
| Current portion of long-term debt |
|
$ |
4,166 |
|
|
$ |
4,166 |
|
|
|
|
| Long-term debt, net of current portion |
|
$ |
1,526 |
|
|
$ |
1,526 |
|
|
|
|
| Stockholders’ equity |
|
|
|
|
|
|
|
|
| Preferred stock, par value $0.0001 per share; 5,000,000 shares authorized, no shares issued and outstanding actual and as
adjusted |
|
|
— |
|
|
|
— |
|
| Common stock, par value $0.0001 per share; 200,000,000 shares authorized, 162,575,446 shares issued and 161,900,446 shares issued and
outstanding, actual; 182,348,173 shares issued and 181,679,480 shares issued and outstanding as adjusted |
|
$ |
15 |
|
|
$ |
17 |
|
| Additional paid-in capital |
|
$ |
295,956 |
|
|
$ |
316,099 |
|
| Accumulated deficit |
|
|
(259,705 |
) |
|
|
(259,705 |
) |
| Less treasury shares at cost, 675,000 shares |
|
|
(3,849 |
) |
|
|
(3,849 |
) |
|
|
|
| Total stockholders’ equity |
|
$ |
32,417 |
|
|
$ |
52,562 |
|
|
|
|
|
|
|
|
|
|
| Total capitalization |
|
$ |
38,109 |
|
|
$ |
58,254 |
|
|
|
|
|
|
|
|
|
|
S-27
PRICE RANGE OF OUR COMMON STOCK
Our common stock is listed on The NASDAQ Capital Market under the symbol “GALE.” The following table shows the high and low per
share sale prices of our common stock for the periods indicated:
|
|
|
|
|
|
|
|
|
| |
|
High |
|
|
Low |
|
| 2014 |
|
|
|
|
|
|
|
|
| First Quarter |
|
$ |
7.77 |
|
|
$ |
2.15 |
|
| Second Quarter |
|
|
3.58 |
|
|
|
1.66 |
|
| Third Quarter |
|
|
3.36 |
|
|
|
2.00 |
|
| Fourth Quarter |
|
|
2.26 |
|
|
|
1.48 |
|
|
|
|
| 2015 |
|
|
|
|
|
|
|
|
| First Quarter |
|
$ |
2.12 |
|
|
$ |
1.33 |
|
| Second Quarter |
|
|
2.39 |
|
|
|
1.27 |
|
| Third Quarter |
|
|
1.92 |
|
|
|
1.10 |
|
| Fourth Quarter |
|
|
1.83 |
|
|
|
1.37 |
|
|
|
|
| 2016 |
|
|
|
|
|
|
|
|
| First Quarter (through January 6, 2016) |
|
$ |
1.48 |
|
|
$ |
1.29 |
|
On January 6, 2016, the last reported sale price of our common stock on The NASDAQ Capital Market was $1.29
per share. On January 6, 2016, there were approximately 544 holders of record of our common stock. The number of holders of record does not include shares held in “street name” through brokers.
DESCRIPTION OF OUR SECURITIES
We are offering 19,772,727 units, consisting of an aggregate of 19,772,727 shares of common stock and warrants to purchase an aggregate of
11,863,636 shares of common stock. Each unit consists of one share of common stock and 0.6 of a warrant to purchase one share of common stock at an exercise price of $1.42 per whole share. The units will not be issued or certificated. The shares of
common stock and the warrants are immediately separable and will be issued separately. This prospectus supplement also relates to the offering of the shares of common stock issuable upon exercise of the warrants being offered in this offering.
Common Stock
As of January 6, 2016,
161,906,753 shares of our common stock were issued and outstanding.
Each holder of our common stock is entitled to one vote for each
share of common stock held on all matters submitted to a vote of stockholders. Common stockholders are not entitled to cumulative voting in the election of directors by our certificate of incorporation. This means that the holders of a majority of
the shares voted will be able to elect all of the directors then standing for election. Subject to preferences that may apply to shares of preferred stock outstanding at the time, the holders of outstanding shares of our common stock are entitled to
receive dividends out of assets legally available at the times and in the amounts that our board of directors may determine from time to time.
Upon our liquidation, dissolution or winding-up, the holders of our common stock will be entitled to share ratably in all assets remaining
after payment of all liabilities and the liquidation preferences of any series of capital stock ranking senior to the common stock upon liquidation. Holders of common stock have no preemptive or conversion rights or other subscription rights. There
are no redemption or sinking fund provisions applicable to the common stock. All outstanding shares of common stock are fully paid and non-assessable, and the shares of common stock to be issued under this prospectus supplement, when they are paid
for, will be fully paid and non-assessable.
Our common stock is listed on The NASDAQ Capital Market under the symbol “GALE.”
The transfer agent of our common stock is Computershare Trust Company, N.A.
Warrants
Form. The warrants will be issued under a warrant agreement to be entered into between us and the warrant agent.
S-28
Exercisability. The warrants will be exercisable upon issuance and will expire on the
five-year anniversary of issuance. The warrants will be exercisable, at the option of each holder, in whole or in part by delivering to us a duly executed exercise notice and payment in full of the exercise price within three Trading Days in
available funds for the number of shares of common stock purchased upon such exercise. If a registration statement registering the issuance or the resale of the shares of common stock underlying the warrants under the Securities Act is not effective
or available, the holder may, in its sole discretion, elect to exercise the warrant through a cashless exercise, in which case the holder would receive upon such exercise the net number of shares of common stock determined according to the formula
set forth in the warrant. No fractional shares of common stock will be issued in connection with the exercise of a warrant. In lieu of fractional shares, we will pay the holders an amount in cash equal to the fractional amount multiplied by the
current market price of our common stock.
Exercise Limitation. A holder will not have the right to exercise any portion of
the warrant if the holder (together with affiliates) would beneficially own in excess of 4.99% of the number of shares of our common stock outstanding immediately after giving effect to the exercise, as such percentage of ownership is determined in
accordance with the terms of the warrants. However, any holder may increase or decrease such percentage to any other percentage not in excess of 9.99% upon at least 61 days’ prior notice from the holder to us.
Exercise Price. The exercise price per whole share of common stock purchasable upon exercise of the warrants is $1.42. The exercise
price is subject to adjustment in the event of certain stock dividends and distributions, stock splits, stock combinations, reclassifications or similar events affecting our common stock and also upon any distribution of assets, including cash,
stock or other property, to our stockholders.
Transferability. Subject to applicable laws, the warrants may be offered for
sale, sold, transferred or assigned without our consent.
Exchange Listing. The warrants will not be listed on The NASDAQ
Capital Market or other securities exchange or nationally recognized trading system.
Fundamental Transactions. In the
event of a fundamental transaction, as described in the warrants and generally including any merger or consolidation with or into another entity, as a result of which the holders of our outstanding voting securities as of immediately prior to such
merger or consolidation hold less than a majority of the outstanding voting securities of the surviving or successor entity as of immediately after such merger or consolidation or a sale, transfer or other disposition of all or substantially all our
property, assets or business to another person or entity, the holders of the warrants will be entitled to receive upon exercise of the warrants the kind and amount of securities, cash or other property that the holders would have received had they
exercised the warrant immediately prior to such fundamental transactions.
In addition in the event of any fundamental transaction
the holder of the warrant has the right, in lieu of receiving the consideration described in the preceding paragraph, to require us to purchase the warrant for an amount of cash backed on the value of the remaining unexercised portion of the warrant
determined in accordance with the Black Scholes option pricing model.
Pro Rata Distributions. In the event that we distribute
debt, securities, rights or warrants to purchase securities or other assets to holders of common stock, then upon exercise of the warrants, the holders will be entitled to receive the same distribution they would have received had they exercised the
warrants immediately prior to the distribution.
Rights as a Stockholder. Except as otherwise provided in the warrants or
by virtue of such holders’ ownership of shares of our common stock, the holder of a warrant does not have the rights or privileges of a holder of our common stock, including any voting rights, until the holder exercises the warrant.
Warrant Agent. Computershare Trust Company, N.A. will act as our warrant agent for the warrants.
S-29
UNDERWRITING
We have entered into an underwriting agreement with the underwriters named below. Raymond James & Associates, Inc., or Raymond James,
is acting as the sole book-running manager and representative of the underwriters. The underwriting agreement provides for the purchase of a specific number of units comprised of shares of common stock and warrants to purchase common stock by each
of the underwriters. The underwriters’ obligations are several, which means that each underwriter is required to purchase a specified number of units, but is not responsible for the commitment of any other underwriter to purchase units. Subject
to the terms and conditions of the underwriting agreement, each underwriter has severally agreed to purchase the number of units set forth opposite its name below:
|
|
|
|
|
| Underwriter |
|
Number of Units |
|
| Raymond James & Associates, Inc. |
|
|
15,323,863 |
|
| Roth Capital Partners, LLC |
|
|
1,482,955 |
|
| Noble Financial Capital Markets |
|
|
1,482,955 |
|
| FBR Capital Markets & Co. |
|
|
988,636 |
|
| Maxim Group LLC |
|
|
494,318 |
|
|
|
|
|
|
| Total |
|
|
19,772,727 |
|
|
|
|
|
|
The underwriters have agreed to purchase all of the units offered by this prospectus supplement (other than
those covered by the over-allotment option described below) if any are purchased.
The shares of common stock and the warrants to purchase
common stock offered hereby should be ready for delivery on or about January 12, 2016 against payment in immediately available funds.
The
underwriters are offering the units subject to various conditions and may reject all or part of any order. The representative of the underwriters has advised us that the underwriters propose to offer the units directly to the public at the public
offering price that appears on the cover page of this prospectus supplement. After the units are released for sale to the public, the representative may change the offering price and other selling terms at various times.
We have granted the underwriters an over-allotment option. This option, which is exercisable for up to 30 days after the date of this
prospectus supplement, permits the underwriters to purchase up to 2,965,909 shares of common stock at a price of $1.0246 per share and/or warrants to purchase up to 1,779,545 shares of common stock at a price of $0.0094 per warrant from us to cover
over-allotments, if any. If this option is exercised in full, the total gross proceeds will be $25 million, and the total net proceeds to us will be $23.5 million. The underwriters have severally agreed that, to the extent the over-allotment option
is exercised, they will each purchase a number of additional units proportionate to the underwriter’s initial amount reflected in the table, above.
The following table provides information regarding the amount of the discounts and commissions to be paid to the underwriters by us, before
expenses:
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Per Unit |
|
|
Total Without
Exercise of Over-
Allotment Option |
|
|
Total With Full
Exercise of Over-
Allotment Option |
|
| Public offering price |
|
$ |
1.10 |
|
|
$ |
21,750,000 |
|
|
$ |
25,012,500 |
|
| Underwriting discounts and commissions |
|
$ |
0.066 |
|
|
$ |
1,305,000 |
|
|
$ |
1,500,750 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Proceeds, before expenses to us |
|
$ |
1.034 |
|
|
$ |
20,445,000 |
|
|
$ |
23,511,750 |
|
We estimate that our total expenses of the offering, excluding the underwriting discounts and commissions,
will be approximately $300,000, which includes $125,000 that we have agreed to reimburse the underwriters for the fees and expenses incurred by them in connection with the offering.
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act of 1933.
We and our officers and directors have agreed to a 90-day “lock-up” with respect to shares of our common stock and other of our
securities that they beneficially own, including securities that are convertible into shares of common stock and securities that are exchangeable or exercisable for shares of common stock. This means that, subject to certain exceptions, for a period
of 90 days following the date of this prospectus supplement, we and such persons may not offer, sell, pledge or otherwise dispose of these securities without the prior written consent of Raymond James. We also have agreed that, without the prior
written consent of Raymond James, for a period of 120 days following the date of this prospectus supplement, we will not sell any shares of common stock pursuant to our purchase agreement with Lincoln Park Capital Fund, LLC.
Rules of the SEC may limit the ability of the underwriters to bid for or purchase shares before the distribution of the shares is completed.
However, the underwriters may engage in the following activities in accordance with the rules:
| |
• |
|
Stabilizing transactions—The representative may make bids or purchases for the purpose of pegging, fixing or maintaining the price of the shares, so long as stabilizing bids do not exceed a specified maximum.
|
| |
• |
|
Over-allotments and syndicate covering transactions—The underwriters may sell more shares of our common stock in connection with this offering
than the number of shares that they have committed to purchase. This over-allotment creates a short position for the underwriters. This short sales position may involve either “covered” short
|
S-30
| |
sales or “naked” short sales. Covered short sales are short sales made in an amount not greater than the underwriters’ over-allotment option to purchase additional shares in this
offering described above. The underwriters may close out any covered short position either by exercising its over-allotment option or by purchasing shares in the open market. To determine how they will close the covered short position, the
underwriters will consider, among other things, the price of shares available for purchase in the open market, as compared to the price at which they may purchase shares through the over-allotment option. Naked short sales are short sales in excess
of the over-allotment option. The underwriters must close out any naked short position by purchasing shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that, in the open market after
pricing, there may be downward pressure on the price of the shares that could adversely affect investors who purchase shares in this offering. |
| |
• |
|
Penalty bids—If the representative purchases shares in the open market in a stabilizing transaction or syndicate covering transaction, it may reclaim a selling concession from the underwriters and selling group
members who sold those shares as part of this offering. |
| |
• |
|
Passive market making—Market makers in the shares who are underwriters or prospective underwriters may make bids for or purchases of shares, subject to limitations, until the time, if ever, at which a stabilizing
bid is made. |
Similar to other purchase transactions, the underwriters’ purchases to cover the syndicate short sales or
to stabilize the market price of our common stock may have the effect of raising or maintaining the market price of our common stock or preventing or mitigating a decline in the market price of our common stock. As a result, the price of the shares
of our common stock may be higher than the price that might otherwise exist in the open market. The imposition of a penalty bid might also have an effect on the price of the shares if it discourages resales of the shares.
Neither we nor the underwriters make any representation or prediction as to the effect that the transactions described above may have on the
price of our common stock. These transactions may occur on The NASDAQ Capital Market or otherwise. If such transactions are commenced, they may be discontinued without notice at any time.
Electronic Delivery of Prospectus Supplement: A prospectus supplement in electronic format may be delivered to potential investors by one or
more of the underwriters participating in this offering. The prospectus supplement in electronic format will be identical to the paper version of such preliminary prospectus supplement. Other than the prospectus supplement in electronic format, the
information on any underwriter’s website and any information contained in any other website maintained by an underwriter is not part of this prospectus supplement, the accompanying prospectus or the registration statement of which this
prospectus supplement and the accompanying prospectus form a part.
NOTICE TO NON-U.S. INVESTORS
Investors are advised to contact their legal, financial or tax advisers to obtain an independent assessment of the financial and tax
consequences of an investment in units.
BELGIUM
The offering is exclusively conducted under applicable private placement exemptions and therefore it has not been and will not be notified to,
and this document or any other offering material relating to the units has not been and will not be approved by, the Belgian Banking, Finance and Insurance Commission (“Commission bancaire, financière et des assurances/Commissie voor het
Bank, Financie en Assurantiewezen”). Any representation to the contrary is unlawful.
Each underwriter has undertaken not to offer
sell, resell, transfer or deliver directly or indirectly, any units, or to take any steps relating/ancillary thereto, and not to distribute or publish this document or any other material relating to the units or to the offering in a manner which
would be construed as: (a) a public offering under the Belgian Royal Decree of 7 July 1999 on the public character of financial transactions; or (b) an offering of securities to the public under Directive 2003/71/EC which triggers an
obligation to publish a prospectus in Belgium. Any action contrary to these restrictions will cause the recipient and us to be in violation of the Belgian securities laws.
FRANCE
Neither this prospectus
supplement nor any other offering material relating to the units has been submitted to the clearance procedures of the Autorité des marchés financiers in France. The units have not been offered or sold and will not be offered or sold,
directly or indirectly, to the public in France. Neither this prospectus supplement nor any other offering material relating to the units
S-31
has been or will be: (a) released, issued, distributed or caused to be released, issued or distributed to the public in France; or (b) used in connection with any offer for subscription
or sale of the units to the public in France. Such offers, sales and distributions will be made in France only: (i) to qualified investors (investisseurs qualifiés) and/or to a restricted circle of investors (cercle restreint
d’investisseurs), in each case investing for their own account, all as defined in and in accordance with Articles L.411-2, D.411-1, D.411-2, D.734-1, D.744-1, D.754-1 and D.764-1 of the French Code monétaire et financier; (ii) to
investment services providers authorised to engage in portfolio management on behalf of third parties; or (iii) in a transaction that, in accordance with article L.411-2-II-1°-or-2°-or 3° of the French Code monétaire et
financier and article 211-2 of the General Regulations (Règlement Général) of the Autorité des marchés financiers, does not constitute a public offer (appel public à l’épargne). Such units may be
resold only in compliance with Articles L.411-1, L.411-2, L.412-1 and L.621-8 through L.621-8-3 of the French Code monétaire et financier.
UNITED KINGDOM / GERMANY / NORWAY / THE NETHERLANDS
In relation to each Member State of the European Economic Area which has implemented the Prospectus Directive (each, a “Relevant Member
State”) an offer to the public of any units which are the subject of the offering contemplated by this prospectus supplement may not be made in that Relevant Member State other than the offers contemplated in this prospectus supplement in
name(s) of Member State(s) where prospectus will be approved or passported for the purposes of a non-exempt offer once this prospectus supplement has been approved by the competent authority in such Member State and published and passported in
accordance with the Prospectus Directive as implemented in name(s) of relevant Member State(s) except that an offer to the public in that Relevant Member State of any units may be made at any time under the following exemptions under the Prospectus
Directive, if they have been implemented in that Relevant Member State:
(a) to legal entities which are authorised or regulated to
operate in the financial markets or, if not so authorised or regulated, whose corporate purpose is solely to invest in securities;
(b) to
any legal entity which has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000 and (3) an annual net turnover of more than €50,000,000,
as shown in its last annual or consolidated accounts;
(c) by the representative to fewer than 100 natural or legal persons (other than
qualified investors as defined in the Prospectus Directive); or
(d) in any other circumstances falling within Article 3(2) of the
Prospectus Directive, provided that no such offer of units shall result in a requirement for the publication by the Company or any underwriter of a prospectus pursuant to Article 3 of the Prospectus Directive.
For the purposes of this provision, the expression an “offer to the public” in relation to any units in any Relevant Member State
means the communication in any form and by any means of sufficient information on the terms of the offer and any units to be offered so as to enable an investor to decide to purchase any units, as the same may be varied in that Member State by any
measure implementing the Prospectus Directive in that Member State and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each Relevant Member State.
This prospectus supplement and any other material in relation to the units is only being distributed to, and is only directed at, persons in
the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospective Directive (“qualified investors”) that also (i) have professional experience in matters relating to investments falling within
Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005, as amended, or the Order, (ii) who fall within Article 49(2)(a) to (d) of the Order or (iii) to whom it may otherwise lawfully be
communicated (all such persons together being referred to as “relevant persons”). The units are only available to, and any invitation, offer or agreement to purchase or otherwise acquire such units will be engaged in only with, relevant
persons. This prospectus supplement and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other person in the United Kingdom. Any person in the United Kingdom
that is not a relevant person should not act or rely on this prospectus supplement or any of its contents.
ISRAEL
In the State of Israel, the units offered hereby may not be offered to any person or entity other than the following:
(a) a fund for joint investments in trust (i.e., mutual fund), as such term is defined in the Law for Joint Investments in Trust, 5754-1994,
or a management company of such a fund;
S-32
(b) a provident fund as defined in Section 47(a)(2) of the Income Tax Ordinance of the State
of Israel, or a management company of such a fund;
(c) an insurer, as defined in the Law for Oversight of Insurance Transactions,
5741-1981, (d) a banking entity or satellite entity, as such terms are defined in the Banking Law (Licensing), 5741-1981, other than a joint services company, acting for their own account or for the account of investors of the type listed in
Section 15A(b) of the Securities Law 1968;
(d) a company that is licensed as a portfolio manager, as such term is defined in
Section 8(b) of the Law for the Regulation of Investment Advisors and Portfolio Managers, 5755-1995, acting on its own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(e) a company that is licensed as an investment advisor, as such term is defined in Section 7(c) of the Law for the Regulation of
Investment Advisors and Portfolio Managers, 5755-1995, acting on its own account;
(f) a company that is a member of the Tel Aviv Stock
Exchange, acting on its own account or for the account of investors of the type listed in Section 15A(b) of the Securities Law 1968;
(g) an underwriter fulfilling the conditions of Section 56(c) of the Securities Law, 5728-1968;
(h) a venture capital fund (defined as an entity primarily involved in investments in companies which, at the time of investment, (i) are
primarily engaged in research and development or manufacture of new technological products or processes and (ii) involve above-average risk);
(i) an entity primarily engaged in capital markets activities in which all of the equity owners meet one or more of the above criteria; and
(j) an entity, other than an entity formed for the purpose of purchasing units in this offering, in which the shareholders equity
(including pursuant to foreign accounting rules, international accounting regulations and U.S. generally accepted accounting rules, as defined in the Securities Law Regulations (Preparation of Annual Financial Statements), 1993) is in excess of NIS
250 million.
Any offeree of the units offered hereby in the State of Israel shall be required to submit written confirmation that it
falls within the scope of one of the above criteria. This prospectus supplement will not be distributed or directed to investors in the State of Israel who do not fall within one of the above criteria.
ITALY
The offering of the units
offered hereby in Italy has not been registered with the Commissione Nazionale per la Società e la Borsa (“CONSOB”) pursuant to Italian securities legislation and, accordingly, the units offered hereby cannot be offered, sold or
delivered in the Republic of Italy (“Italy”) nor may any copy of this prospectus supplement or any other document relating to the units offered hereby be distributed in Italy other than to professional investors (operatori qualificati) as
defined in Article 31, second paragraph, of CONSOB Regulation No. 11522 of 1 July, 1998 as subsequently amended. Any offer, sale or delivery of the units offered hereby or distribution of copies of this prospectus supplement or any other
document relating to the units offered hereby in Italy must be made:
(a) by an investment firm, bank or intermediary permitted to conduct
such activities in Italy in accordance with Legislative Decree No. 58 of 24 February 1998 and Legislative Decree No. 385 of 1 September 1993 (the “Banking Act”);
(b) in compliance with Article 129 of the Banking Act and the implementing guidelines of the Bank of Italy; and
(c) in compliance with any other applicable laws and regulations and other possible requirements or limitations which may be imposed by
Italian authorities.
SWEDEN
This prospectus supplement has not been nor will it be registered with or approved by Finansinspektionen (the Swedish Financial Supervisory
Authority). Accordingly, this prospectus supplement may not be made available, nor may the units offered hereunder be marketed and offered for sale in Sweden, other than under circumstances which are deemed not to require a prospectus under the
Financial Instruments Trading Act (1991: 980).
S-33
SWITZERLAND
The units being offered pursuant to this prospectus supplement will not be offered, directly or indirectly, to the public in Switzerland and
this prospectus supplement does not constitute a public offering prospectus as that term is understood pursuant to art. 652a or art. 1156 of the Swiss Federal Code of Obligations. We have not applied for a listing of the units being offered pursuant
to this prospectus supplement on the SWX Swiss Exchange or on any other regulated securities market, and consequently, the information presented in this prospectus supplement does not necessarily comply with the information standards set out in the
relevant listing rules. The units being offered pursuant to this prospectus supplement have not been registered with the Swiss Federal Banking Commission as foreign investment funds, and the investor protection afforded to acquirers of investment
fund certificates does not extend to acquirers of units.
LEGAL MATTERS
TroyGould PC, Los Angeles, California, has rendered an opinion with respect to the validity of the securities offered by this prospectus
supplement. Sanford J. Hillsberg, the Chairman of our board of directors, is an attorney with TroyGould PC. TroyGould PC and some of its attorneys own shares of our common stock constituting in the aggregate less than 1% of our outstanding shares of
common stock. The underwriters are being represented in connection with this offering by Mintz, Levin, Cohn, Ferris, Glovsky, and Popeo, P.C., New York, New York.
EXPERTS
The consolidated financial statements of the company as of December 31, 2014 and 2013, and for the years then ended, and the effectiveness of
internal control over financial reporting of the company as of December 31, 2014, have been incorporated by reference herein in reliance upon the reports of Moss Adams LLP, independent registered public accounting firm, incorporated by reference
herein, and upon the authority of said firm as experts in accounting and auditing. The consolidated financial statements of the company as of December 31, 2012 and for the year then ended incorporated in this prospectus supplement by reference to
our Annual Report on Form 10-K for the year ended December 31, 2014, have been so incorporated in reliance on the report of BDO USA, LLP, an independent registered public accounting firm, upon the authority of said firm as experts in auditing and
accounting.
WHERE YOU CAN FIND MORE INFORMATION
We have filed a registration statement on Form S-3 with the SEC under the Securities Act of 1933. This prospectus supplement is part of the
registration statement, but the registration statement includes and incorporates by reference additional information and exhibits. We file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and
copy the registration statement and any other document we file with the SEC at the public reference room maintained by the SEC at 100 F Street, N.E., Washington, D.C. 20549. You may obtain information on the operation of the public reference room by
calling the SEC at 1-800-SEC-0330. The SEC also maintains a web site that contains reports, proxy and information statements and other information regarding companies, such as ours, that file documents electronically with the SEC. The address of
that site on the world wide web is http://www.sec.gov. The information on the SEC’s web site is not part of this prospectus supplement, and any references to this web site or any other web site are inactive textual references only.
S-34
INCORPORATION OF DOCUMENTS BY REFERENCE
The SEC permits us to “incorporate by reference” the information contained in documents we file with the SEC, which means that we
can disclose important information to you by referring you to those documents rather than by including them in this prospectus supplement or the accompanying prospectus. Information that is incorporated by reference is considered to be part of this
prospectus supplement, and you should read it with the same care that you read this prospectus supplement. Later information that we file with the SEC will automatically update and supersede the information that is either contained, or incorporated
by reference, in this prospectus supplement, and will be considered to be a part of this prospectus supplement from the date those documents are filed.
We incorporate by reference into this prospectus supplement the following documents and information filed with the SEC:
| |
• |
|
our Annual Report on Form 10-K for the year ended December 31, 2014, which was filed on March 5, 2015, and amended on March 10, 2015 (Items 6, 7, and 8 from said Annual Report have been recast for discontinued
operations as reflected in the Current Report on Form 8-K filed on December 4, 2015); |
| |
• |
|
our Quarterly Report on Form 10-Q for the three months ended March 31, 2015 filed with the SEC on May 7, 2015; |
| |
• |
|
our Quarterly Report on Form 10-Q for the three months ended June 30, 2015 filed with the SEC on August 6, 2015; |
| |
• |
|
our Quarterly Report on Form 10-Q for the three months ended September 30, 2015 filed with the SEC on November 9, 2015; |
| |
• |
|
our Current Reports on Form 8-K filed with the SEC on January 14, 2015, February 9, 2015, March 5, 2015, March 16, 2015, April 9, 2015, June 24,
2015, October 27, 2015, October 30, 2015, November 20, 2015, December 4, 2015, December 11, 2015, December 18, 2015, December 22, 2015, December 24, 2015, December 29,
2015 and January 7, 2016, respectively; and |
| |
• |
|
the description of our common stock and related rights contained in our registration statement on Form 8-A (File No. 001-33958), including any amendment or report filed for the purpose of updating such description.
|
We also incorporate by reference into this prospectus supplement all additional documents that we file with the SEC under
the terms of Section 13(a), 13(c), 14 or 15(d) of the Securities Exchange Act of 1934 that are made after the date of this prospectus supplement and before the termination of the offering of securities offered by this prospectus supplement. We
are not, however, incorporating, in each case, any documents or information that we are deemed to furnish and not file in accordance with SEC rules.
You may request a copy of any of the documents incorporated by reference into this prospectus supplement, at no cost, by writing or
telephoning us at the following address: Galena Biopharma, Inc., 2000 Crow Canyon Place, Suite 380, San Ramon, California 94583, Attention: Investor Relations, Phone: (855) 855-4253. We will not send exhibits to any documents, unless the
exhibits are specifically incorporated by reference into the document.
S-35
PROSPECTUS

GALENA BIOPHARMA, INC.
$100,000,000
Common
Stock
Preferred Stock
Warrants
Rights
Units
We may, from
time to time, offer and sell shares of common stock, shares of preferred stock, warrants or rights, either separately or in units, in one or more offerings. The preferred stock and warrants may be convertible into or exercisable or exchangeable for
common stock or preferred stock. The rights may be exercisable for common stock or preferred stock. We will specify in the accompanying prospectus supplement more specific information about any such offering. The aggregate initial offering price of
all securities sold under this prospectus will not exceed $100,000,000, including the U.S. dollar equivalent if the public offering of any such securities is denominated in one or more foreign currencies, foreign currency units or composite
currencies.
We may offer these securities for sale directly to investors or through underwriters, dealers or agents. We will set forth
the names of any underwriters, dealers or agents and their compensation in the accompanying prospectus supplement.
This prospectus may
not be used to sell any of these securities unless accompanied by a prospectus supplement.
Our common stock is traded on The NASDAQ
Capital Market under the symbol “GALE.” On December 3, 2015, the last reported sale price of our common stock on The NASDAQ Capital Market was $1.45 per share.
Investing in our securities involves risks. See the section entitled “Risk Factors” in the
accompanying prospectus supplement and in the documents we incorporate by reference in this prospectus.
NEITHER THE SECURITIES AND
EXCHANGE COMMISSION NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED OF THESE SECURITIES OR PASSED UPON THE ADEQUACY OR ACCURACY OF THIS PROSPECTUS. ANY REPRESENTATION TO THE CONTRARY IS A CRIMINAL OFFENSE.
The date of this prospectus is December 22, 2015
Prospectus No. 1
TABLE OF CONTENTS
ABOUT THIS PROSPECTUS
This prospectus is part of a registration statement that we filed with the Securities and Exchange Commission, or “SEC,” using a
“shelf” registration, or continuous offering, process. Under this shelf registration process, we may, from time to time, offer and sell shares of common stock, shares of preferred stock, warrants or rights, either separately or in units,
in one or more offerings with a maximum aggregate offering price of $100,000,000.
This prospectus provides you with a general description
of the securities we may offer. Each time we sell securities, we will provide a prospectus supplement that will contain specific information about the terms of that offering and the offered securities. Any prospectus supplement may also add, update
or change information contained in this prospectus. Any statement that we make in this prospectus will be modified or superseded by any inconsistent statement made by us in a prospectus supplement. The registration statement we filed with the SEC
includes exhibits that provide more detail of the matters discussed in this prospectus. You should read this prospectus and the related exhibits filed with the SEC and any prospectus supplement, together with additional information described under
the heading “Where You Can Find More Information,” before making your investment decision.
Unless otherwise indicated,
information contained or incorporated by reference in this prospectus concerning our industry, including our general expectations and market opportunity, is based on information from our own management estimates and research, as well as from
industry and general publications and research, surveys and studies conducted by third parties. Management estimates are derived from publicly available information, our knowledge of our industry and assumptions based on such information and
knowledge, which we believe to be reasonable. In addition, assumptions and estimates of our and our industry’s future performance are necessarily uncertain due to a variety of factors, including those referred to in “Risk Factors”
below in this prospectus. These and other factors could cause our future performance to differ materially from our assumptions and estimates.
NeuVax™ is our trademark used in this prospectus. This prospectus also includes the
Zuplenz® trademark and Abstral® and other trademarks, trade names and service marks that are the property of other organizations but in
certain instances have been licensed to us. Solely for convenience, trademarks and trade names referred to in this prospectus sometimes appear without the ® and ™ symbols, but those
references are not intended to indicate that we will not assert, to the fullest extent under applicable law, our rights, or that the applicable owner will not assert its rights, to these trademarks and trade names.
NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and the other documents we have filed with the SEC that are incorporated herein by reference contain forward-looking
statements within the meaning of the Private Securities Litigation Reform Act of 1995 that involve risks and uncertainties, as well as assumptions that, if they never materialize or prove incorrect, could cause our results to differ materially from
those expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be deemed forward-looking statements, including any projections of financing needs, revenue, expenses,
earnings or losses from operations, or other financial items, any statements of the plans, strategies and objectives of management for future operations, any statements concerning product research, development and commercialization plans and
timelines, any statements regarding safety and efficacy of product candidates, any statements of expectation or belief and any statements of assumptions underlying any of the foregoing. In addition, forward-looking statements may contain the words
“believe,” “anticipate,” “expect,” “estimate,” “intend,” “plan,” “project,” “will be,” “will continue,” “will result,” “seek,”
“could,” “may,” “might,” or any variations of such words or other words with similar meanings. All forward-looking statements attributable to us or to persons acting on our behalf are expressly qualified in their
entirety by the cautionary statements and risk factors set forth under “Risk Factors” and elsewhere in this prospectus and set forth in our Form 10-K for the year ended December 31, 2014 and subsequent Quarterly Reports on Form 10-Q
filed with the SEC. See “Where You Can Find More Information” and “Incorporation of Certain Documents by Reference” in this prospectus for information on how to access or obtain our reports filed with the SEC.
Given their inherent uncertainty, you should not place undue reliance on these forward-looking statements. You should read this prospectus and
the documents that we reference in this prospectus with the understanding that our actual future results may be materially different from what we expect. Except as required by law, we do not undertake any obligation to update or revise any
forward-looking statements contained in this prospectus, whether as a result of new information, future events or otherwise.
RISK FACTORS
Investing in our securities involves significant risks. The prospectus supplement
relating to a particular offering will contain a discussion of risks applicable to an investment in the securities offered. Prior to making a decision about investing in our securities, you should carefully consider the specific factors discussed
under the heading “Risk Factors” in the applicable prospectus supplement together with all of the other information contained in the prospectus supplement or appearing or incorporated by reference in this prospectus.
1
Prospectus No. 1
ABOUT GALENA
Overview
Galena Biopharma, Inc.
(“we,” “us,” “our,” “Galena” or the “company”) is a biopharmaceutical company committed to the development and commercialization of targeted oncology therapeutics that address major unmet medical
needs. Galena’s development portfolio is focused primarily on addressing the rapidly growing patient populations of cancer survivors by harnessing the power of the immune system to prevent cancer recurrence. The Company’s pipeline consists
of multiple mid- to late-stage clinical assets, including novel cancer immunotherapy programs led by NeuVax™ (nelipepimut-S) and GALE-301. NeuVax is currently in a pivotal, Phase 3 clinical trial with several concurrent Phase 2 trials ongoing
both as a single agent and in combination with other therapies. GALE-301 is in a Phase 2a clinical trial in ovarian and endometrial cancers and in a Phase 1b given sequentially with GALE-302.
We are seeking to build value for shareholders through pursuit of the following objectives:
| |
• |
|
Develop novel cancer immunotherapies to address unmet medical needs through the use of peptide-based vaccines targeting well-established tumor antigens. One of our key strategies is to target the adjuvant, minimum
residual disease setting, in high risk patients who are more likely to benefit from treatment via immunotherapy. Our immunotherapy programs are currently targeting two key areas: secondary prevention to seek to significantly decrease the risk of
disease recurrence in breast cancer, gastric cancer, endometrial and ovarian cancers; and a planned trial positioning earlier in the breast cancer treatment spectrum via primary prevention. |
| |
• |
|
Expand our development pipeline by enhancing the clinical and geographic footprint of our technologies. We can accomplish this through the initiation of new clinical trials as well as through acquisition of additional
development stage products in related oncology indications. |
| |
• |
|
Leverage valuable partnerships and collaborations, as well as investigator-sponsored trial arrangements, to maximize the scope of potential clinical opportunities in a cost effective and efficient manner.
|
| |
• |
|
Focus our resources on our valuable and expanding clinical development programs. On November 19, 2015, we sold our Abstral® (fentanyl) Sublingual Tablets
product and related assets and we have determined to sell or otherwise dispose of our Zuplenz® (ondansetron) Oral Soluble Film product and related assets and to cease our commercial operations
to seek to maximize value to our shareholders. |
The chart below summarizes the current status of our clinical development
pipeline:
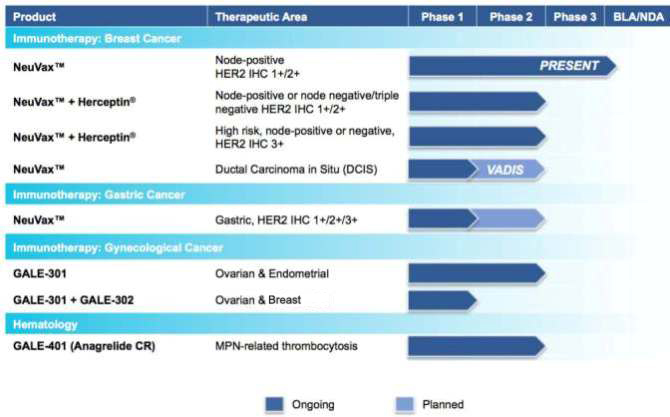
2
Prospectus No. 1
Novel Cancer Immunotherapies
Our targeted cancer immunotherapy approach is currently based upon two key areas: preventing secondary recurrence of cancer, which is becoming
increasingly important as the number of cancer survivors continues to grow; and, positioning earlier in the breast cancer treatment spectrum via primary prevention. Once a patient’s tumor becomes metastatic, the outcome is most often fatal,
making the prevention of recurrence a potentially critical component of overall patient care. Our programs primarily target patients in the adjuvant (after-surgery) setting who have relatively healthy immune systems, but may still have minimal
residual disease. Minimal residual disease, or single cancer cells (occult cancer cells) or micrometastasis, are undetectable by current radiographic scanning technologies, but we know can result in disease recurrence.
Our therapies utilize an immunodominant peptide combined with the immune adjuvant, recombinant human granulocyte macrophage-colony stimulating
factor (rhGM-CSF), and work by harnessing the patient’s own immune system to seek out and attack any residual cancer cells. Using peptide immunogens has many potential clinical advantages, including a favorable safety profile, since these drugs
may lack the toxicities typical of most cancer therapies. They also have the potential to evoke long-lasting protection through activation of the immune system and a convenient, intradermal mode of delivery. We are currently engaged in multiple
clinical trials with NeuVax™ (nelipepimut-S), GALE-301, and GALE-302, targeting the prevention of recurrence in breast, gastric, ovarian and endometrial cancers.
NeuVax™ (nelipepimut-S)
NeuVax™ (nelipepimut-S), our lead product candidate, is a cancer immunotherapy targeting human epidermal growth factor receptor (HER2)
expressing cancers. NeuVax is the immunodominant nonapeptide derived from the extracellular domain of the HER2 protein, a well-established and validated target for therapeutic intervention in breast and gastric carcinomas. The NeuVax vaccine is
combined with GM-CSF for injection under the skin, or intradermal administration. Data has shown that an increased presence of circulating tumor cells (CTCs) may predict Disease Free Survival (DFS) and Overall Survival (OS)—suggesting a
dormancy of isolated micrometastases, which, over time, may lead to recurrence. After binding to the specific HLA molecules on antigen presenting cells, the nelipepimut-S sequence stimulates specific cytotoxic T lymphocyte (CTLs). These activated
CTLs recognize, neutralize and destroy, through cell lysis, HER2 expressing cancer cells, including occult cancer cells and micrometastatic foci. The nelipepimut immune response can also generate CTLs to other immunogenic peptides through inter- and
intra-antigenic epitope spreading.
Breast Cancer: According to the National Cancer Institute, over 230,000 women in the U.S. are
diagnosed with breast cancer annually. While improved diagnostics and targeted therapies have decreased breast cancer mortality in the U.S., metastatic breast cancer remains incurable. Approximately 75% of breast cancer patients have tissue test
positive for some increased amount of the HER2 receptor, which is associated with disease progression and decreased survival. Only approximately 20% to 30% of all breast cancer patients—those with HER2 immunohistochemistry (IHC) 3+ disease, or
IHC 2+ and fluorescence in situ hybridization (FISH) positive—have a HER2 directed, approved treatment option available. This leaves the majority of breast cancer patients with low-to-intermediate HER2 IHC 1+/2+ ineligible for therapy and
without an effective targeted treatment option to prevent cancer recurrence.
We have multiple trials currently ongoing for NeuVax. For
our pivotal, Phase 3 PRESENT (Prevention of Recurrence in Early-Stage, Node- Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) trial, NeuVax is targeting the
30,000-40,000 of the 230,000 female breast cancer patients annually diagnosed in the U.S. who are at a higher risk of their breast cancer recurring, which we refer to as “disease recurrence,” after achieving “no evidence of
disease” (NED) status, (or becoming a “survivor”) with standard-of-care therapy (surgery, chemotherapy, radiation). These high-risk patients have a particular molecular signature and disease status: HER2 IHC 1+/2+ (oncoprotein
associated with aggressive tumor growth), node positive (disease present in the axillary lymph nodes prior to surgery), and HLA A2/A3 (human leukocyte antigen from A2/A3 patients who have the same loci of genes which represents approximately 65% of
the population). Up to 25% of resectable, node-positive breast cancer patients, having no radiographic evidence of disease following surgery and adjuvant chemo/radiation therapy, are expected to relapse within three years following diagnosis. The
prognosis upon recurrence is very poor. These cancer patients presumably still had isolated, undetected tumor CTCs that led to a recurrence of cancer in the breast (local recurrence) or in another location (metastatic disease). In addition to our
Phase 3 trial, we currently have two additional Phase 2 breast cancer trials ongoing with NeuVax in combination with trastuzumab (Herceptin®; Genentech/Roche) targeting the prevention of
recurrence in expanded indications.
We also recently announced our intent to initiate a Phase 2 trial with NeuVax as a single agent in
patients with ductal carcinoma in situ (DCIS) in collaboration with the National Cancer Institute (NCI), potentially positioning NeuVax earlier in the treatment cycle towards primary prevention. The trial will have an immunological endpoint
evaluating NeuVax peptide-specific cytotoxic T lymphocyte (CTL; CD8+ T cell) response in vaccinated patients. DCIS, is defined by the NCI as a noninvasive condition in which abnormal cells are found in the lining of a breast duct, and is the most
common type of breast cancer. The abnormal cells have not spread outside the duct to other tissues in the breast. In some cases, DCIS may become invasive cancer and spread to other tissues, and at this time, there is no way to know which lesions
could become invasive. Current treatment options for DCIS include breast- conserving surgery and radiation therapy with or without tamoxifen, breast-conserving surgery without radiation therapy, or total mastectomy with or without tamoxifen.
According to the American Cancer Society, in 2014 there were an estimated 51,933 diagnoses of DCIS.
3
Prospectus No. 1
Gastric Cancer: Gastric cancer (also known as stomach cancer) is a disease in which the
cells forming the inner lining of the stomach become abnormal and start to divide uncontrollably, forming a cancerous tumor mass. Cancer can develop in any of the five sections of the stomach. Symptoms and outcomes of the disease will vary depending
on the location of the cancer. Stomach cancer is one of the leading causes of cancer deaths in several areas of the world, most notably in Asia. Annually, almost one million people will be diagnosed worldwide with stomach cancer and over 700,000
will die from the disease. More than 90% of stomach cancers are caused by adenocarcinomas, malignant cancers that originate in glandular tissues. Overexpression of the HER2 receptor occurs in approximately 20% of gastric and gastro-esophageal
junction adenocarcinomas, predominantly those of the intestinal type. Overall, without regard to the stage of cancer, only approximately 28% of patients with stomach cancer live at least five years following diagnosis and new adjuvant treatments are
needed to prevent disease recurrence.
We currently have a number of ongoing or planned clinical trials designed to expand the clinical
and geographical footprint of NeuVax:
| |
• |
|
Phase 3 Ongoing: Our Phase 3 PRESENT (Prevention of Recurrence in Early- Stage, Node-Positive Breast Cancer with Low to Intermediate HER2 Expression with NeuVax Treatment) study
targeted enrollment of 700 HER2 1+/2+ patients under a Special Protocol Assessment (SPA) granted by the U.S. Food and Drug Administration (FDA). The multinational, multicenter, randomized, double-blinded PRESENT trial is ongoing in North
America, Western and Eastern Europe, and Israel. The trial is fully enrolled with 758 patients. |
| |
• |
|
Phase 2b Ongoing: A randomized, multicenter, investigator-sponsored, 300 patient Phase 2b clinical trial is enrolling HER2 1+/2+ node-positive and high-risk node-negative breast cancer patients who are HLA A2+, A3+,
A24+ or A26+ to study NeuVax in combination with trastuzumab in the adjuvant setting. This trial is co-funded by Genentech/Roche (providing both trastuzumab and monetary support) and Galena (providing NeuVax and monetary support). |
| |
• |
|
Phase 2 Ongoing: An investigator-sponsored trial is ongoing to study NeuVax in combination with trastuzumab. The study will enroll 100 node positive and negative HER2 IHC 3+ patients or HER2 gene-amplified breast cancer
patients who are HLA A2+ or HLA A3+ and are determined to be at high-risk for recurrence. Partial funding for this trial comes from the Department of Defense (DoD) through the Congressionally Directed Medical Research Program via legislation known
as the Defense Appropriations Act. The grant was awarded under a Breast Cancer Research Program with the Breakthrough Award given to the lead investigator for the trial. |
| |
• |
|
Phase 2 Planned: A clinical trial, entitled, VADIS: Phase 2 trial of the Nelipepimut-S Peptide VAccine in Women with DCIS of the Breast is planned to initiate by the end of 2015/early 2016. The
Phase 2 trial will be a single-blind, double arm, randomized, controlled trial in pre- or post-menopausal patients with DCIS and are HLA-A2 positive. VADIS will be co-funded and run in collaboration with the National Cancer Institute (NCI).
|
| |
• |
|
Phase 2 Planned: A Phase 2 clinical trial in patients with gastric cancer is expected to initiate in 2016. The trial will be run in India by our partner, Dr. Reddy’s Laboratories, Ltd., as part of our NeuVax
commercialization agreement in that region with Dr. Reddy’s. |
GALE-301 and GALE-302
Our second immunotherapy franchise targets folate binding protein receptor-alpha, a well-validated therapeutic target, which is highly
over-expressed (20-80 fold) in ovarian, endometrial and breast cancers. Both GALE-301 (E39) and GALE-302 (E39’) are immunogenic peptides that can stimulate CTLs to recognize and destroy FBP-expressing cancer cells. GALE-301 consists of the FBP
peptide E39 combined with GM-CSF, and is currently in a Phase 2a clinical trial for the prevention of recurrence in patients with ovarian and endometrial cancers. GALE-302 is an attenuated version of the E39 peptide and is currently in a Phase 1b
randomized, single-center trial investigating a novel vaccination series using GALE-301 and GALE-302 to evaluate the immune response and monitor long-term immunity. Current treatments after surgery for these diseases are principally with platinum
based chemotherapeutic agents and patients suffer a high recurrence rate; and, most patients relapse with an extremely poor prognosis. Although not powered for efficacy, promising preliminary results from the Phase 2a clinical trial of GALE-301 were
presented in September 2015 at the European Cancer Congress and demonstrated statistically significant data with the estimate for disease free survival at two years at 85.7% (1000 mcg dose group) vs. 33.6% for the control group (p < .02), and
that GALE-301 was well-tolerated with primarily Grade 1 and 2 toxicities and elicited a strong in vivo immune response.
In November 2015, we presented preliminary data at the Society for Immunotherapy of Cancer Conference on the primary vaccine series (PVS) from
a randomized Phase lb trial with GALE-301 and GALE-302 demonstrating that the in vivo immune response is enhanced with the use of the attenuated E39’ (GALE-302) after E39 (GALE-301). Both agents were shown to be immunogenic and well tolerated
with no differences in toxicities between primary vaccine sequences.
Ovarian and Endometrial Cancer: According to the NCI
Surveillance, Epidemiology, and End Results (SEER) Program, new cases of ovarian cancer occur at an annual rate of 12.1 per 100,000 women in the U.S., with an estimated 21,290 cases for 2015. Although ovarian cancer represents about 1.3% of all
cancers, it represents about 2.4% of all cancer deaths, or an estimated 14,180 deaths in 2015. Approximately 1.3% of women will be diagnosed with ovarian cancer at
4
Prospectus No. 1
some point during their lifetime (2010 – 2012 data). The prevalence of ovarian cancer in the U.S. is about 192,000 women, and the five-year survivorship for women with ovarian cancer is
45.6%. Due to the lack of specific symptoms, the majority of ovarian cancer patients are diagnosed at later stages of the disease, with an estimated 75% of women presenting with advanced-stage (III or IV) disease. These patients have their
tumors routinely surgically debulked to minimal residual disease, and then are treated with platinum- and/or taxane-based chemotherapy. While many patients respond to this treatment regimen and become clinically free-of-disease, the majority of
these patients will relapse. Depending upon their level of residual disease, the risk for recurrence after completion of primary therapy ranges from 60% to 85%. Unfortunately for these women, once the disease recurs, treatment options are limited
and the disease remains incurable.
According to the NCI SEER Program, new cases of endometrial cancer occur at an annual rate of
25.1 per 100,000 women in the U.S., with an estimated 54,870 cases for 2015. Although endometrial cancer represents about 3.3% of all cancers, it represents about 1.7% of all cancer deaths, or an estimated 10,170 deaths in 2015. Approximately
2.8% of women will be diagnosed with endometrial cancer at some point during their lifetime (2010 – 2012 data). The prevalence of endometrial cancer in the U.S. is about 620,000 women, and the five-year survivorship for women with endometrial
cancer is 81.7%.
Hematology
GALE-401 (anagrelide controlled release (CR))
In January 2014, we announced the acquisition of the worldwide rights to anagrelide controlled release (CR), which we renamed GALE-401, through
our acquisition of Mills Pharmaceuticals, LLC. GALE-401 contains the active ingredient anagrelide, an FDA-approved product, for the treatment of patients with myeloproliferative neoplasms (MPNs) to lower abnormally elevated platelet levels. The
currently available immediate release (IR) version of anagrelide causes adverse events that are believed to be dose and plasma concentration dependent. Therefore, reducing the maximum concentration (Cmax) is hypothesized to reduce the side effects,
but preserve efficacy.
Multiple Phase 1 studies in 98 healthy subjects have shown GALE-401 reduces the Cmax of anagrelide following oral
administration, appears to be well-tolerated at the doses administered, and to be capable of reducing platelet levels. The Phase 1 program provided the desired PK/PD (pharmacokinetic/pharmacodynamic) profile to enable the initiation of the ongoing
Phase 2 proof-of-concept trial. The Phase 2, open label, single arm, proof-of-concept trial enrolled 18 patients in the United States for the treatment of thrombocytosis, or elevated platelet counts in patients with MPNs. Phase 2 top-line safety and
efficacy data was presented in June 2015 at the European Hematology Association 20th Congress, and we expect to present a more mature data set at the 57th American Society of Hematology Annual
Meeting in December 2015. Based on a regulatory meeting with the FDA, Galena believes a 505(b)(2) regulatory filing is an acceptable pathway for development and potential approval of GALE-401.
Myeloproliferative neoplasms: MPNs are a closely related group of hematological malignancies in which the bone marrow cells that produce the
body’s blood cells develop and function abnormally. The main MPNs are Polycythemia Vera (PV), Essential Thrombocythemia (ET), Primary Myelofibrosis (PMF), and Chronic Myelogenous Leukemia (CML), all of which are associated with high platelet
counts. The MPNs are progressive blood cancers that can strike anyone at any age, and for which there is no known cure.
Strategic Review of
Commercial Operations
During the quarter ended September 30, 2015, we completed a strategic review of the commercial business
and operations. As a result of the review, we recently sold our Abstral® (fentanyl) Sublingual Tablets product and related assets, and we are pursuing the sale or other disposition of our
Zuplenz® (odansetron) Oral Soluble Film product and related assets described below.
Abstral® (fentanyl) Sublingual Tablets
On November 19, 2015, we and Sentynl Therapeutics Inc., or Sentynl, entered into an asset purchase agreement pursuant to which we sold to
Sentynl our Abstral® (fentanyl) Sublingual Tablets product and related assets, including our rights in the asset purchase agreement and the license agreement between us and Orexo AB dated
March 15, 2013, and March 18, 2013, respectively, and the settlement and license agreement among Orexo AB, Actavis Laboratories FL, Inc, and us dated October 23, 2015. Sentynl has assumed our future obligations under the Orexo agreements,
except that we will continue to be responsible for certain product channel liabilities. We also will continue to be responsible for any pre-closing liabilities and obligations related to Abstral.
Under the purchase agreement we received from Sentynl an $8 million upfront cash payment and are entitled to up to an aggregate of
$4 million in future cash payments based on Sentynl’s achievement of specified Abstral net sales milestones. The purchase agreement also includes customary representations, warranties, covenants and indemnities by Sentynl and us.
Zuplenz® (ondansetron) Oral Soluble Film
Zuplenz® (ondansetron) Oral Soluble Film, is approved by the FDA in adult patients for
the prevention of highly and moderately emetogenic chemotherapy-induced nausea and vomiting (CINV), radiotherapy-induced nausea and vomiting (RINV), and post-operative nausea and vomiting (PONV). Zuplenz is also approved in pediatric patients
treated with moderately emetogenic
5
Prospectus No. 1
CINV. Nausea and vomiting are two of the most common side-effects experienced by post-surgery patients and patients receiving chemotherapy or radiation. Zuplenz utilizes MonoSol’s
proprietary PharmFilm® technology, an oral soluble film that dissolves on the tongue in less than 30 seconds, therefore eliminating the burden of swallowing pills during periods of emesis. The
active pharmaceutical ingredient in Zuplenz, ondansetron, belongs to a class of medications called serotonin 5-HT3 receptor antagonists and works by blocking the action of serotonin, a natural substance that may cause nausea and vomiting.
Alliance Partners in Therapeutic Areas
We are actively looking to leverage our technology platforms by seeking to work with pharmaceutical and biotechnology partners in a number of
therapeutic areas in oncology. Our team has experience targeting products in multiple indications, and based on this experience, we believe we can expand the clinical utility of our current development candidates as well as discover more drug
candidates by working with partners than we can develop with our own resources. We are seeking to work with partners in the discovery and development of drugs in a number of therapeutic areas and technology platforms.
Intellectual Property
Patents and other
intellectual property rights are crucial to our success. It is our policy to protect our intellectual property rights through available means, including filing and prosecuting patent applications in the U.S. and other countries, protecting trade
secrets, and utilizing regulatory protections such as data exclusivity. We also include restrictions regarding use and disclosure of our proprietary information in our contracts with third parties, and utilize customary confidentiality agreements
with our employees, consultants, clinical investigators and scientific advisors to protect our confidential information and know-how. Together with our licensors, we also rely on trade secrets to protect our combined technology especially where we
do not believe patent protection is appropriate or obtainable. It is our policy to operate without infringing on, or misappropriating, the proprietary rights of others. The following chart summarizes our intellectual property rights:
6
Prospectus No. 1
|
|
|
|
|
|
|
|
|
| Drug Candidate |
|
Indication |
|
Scope |
|
Strategic Partner |
|
Estimated
Exclusivity
Period |
| NeuVax™ (nelipepimut-S) |
|
Breast cancer recurrence |
|
Filed and pending or issued
worldwide |
|
University of
Texas/MDACC/
Henry M. Jackson
Foundation |
|
2028 |
|
|
|
|
|
| NeuVax™ (nelipepimut-S) |
|
Gastric |
|
Filed and pending or issued
worldwide |
|
Dr. Reddy’s
Laboratories |
|
2028 |
|
|
|
|
|
| NeuVax™ (nelipepimut-S) |
|
DCIS |
|
Filed and pending or issued
worldwide |
|
National Cancer
Institute/
University of
Texas, MD
Anderson Cancer
Center |
|
2028 |
|
|
|
|
|
| NeuVax™ in combination with Herceptin® |
|
Breast cancer |
|
Filed and pending or issued
worldwide |
|
Henry M. Jackson
Foundation,
Genentech/Roche |
|
2026 |
|
|
|
|
|
| NeuVax™ in combination with other compounds |
|
Breast cancer |
|
Filed and pending or issued
worldwide |
|
Pending |
|
2037 |
|
|
|
|
|
| GALE-301 & GALE-302 |
|
Breast, ovarian and endometrial cancer |
|
Filed and pending or issued
worldwide |
|
Henry M. Jackson
Foundation |
|
2035 |
|
|
|
|
|
| GALE-401 (Anagrelide Controlled Release) |
|
Platelet Lowering |
|
Filed and pending or issued
worldwide |
|
BioVascular, Inc. |
|
2029 |
Out-License Agreements
Teva Pharmaceuticals
Effective December 3, 2012, we entered into a license and supply agreement with ABIC Marketing Limited, a subsidiary of Teva
Pharmaceuticals (“ABIC”). Under the agreement, we granted ABIC exclusive rights to seek marketing approval in Israel for our NeuVax product candidate for the treatment of breast cancer following its approval by the FDA or the European
Medicines Agency, and to market, sell and distribute NeuVax in Israel assuming such approval is obtained. ABIC’s rights also include a right of first refusal in Israel for all future indications for which NeuVax may be approved.
Under the license and supply agreement, ABIC will assume responsibility for regulatory registration of NeuVax in Israel, provide financial
support for local development, and commercialize the product in the region in exchange for making royalty payments to us based on future sales of NeuVax. ABIC also agrees in the license and supply agreement to purchase all supplies of NeuVax from us
at a price determined according to a specified formula.
Dr. Reddy’s Laboratories Ltd.
Effective January 14, 2014, we entered into a strategic development and commercialization partnership with Dr. Reddy’s
Laboratories Ltd. (“Dr. Reddy’s”), under which we licensed commercial rights in India to Dr. Reddy’s for NeuVax in breast and gastric cancers. Under the agreement, Dr. Reddy’s will lead the Phase 2 development of
NeuVax in India in gastric cancer, significantly expanding the potential patient population addressable with NeuVax.
Kwangdong
Pharmaceutical Co., Ltd.
Effective April 30, 2009, we entered into a license agreement with Kwangdong Pharmaceutical Co, Ltd
(Kwangdong). Under the agreement, we granted Kwangdong exclusive rights to seek marketing approval in The Republic of Korea (South Korea) for our NeuVax product candidate for the treatment of breast cancer following its approval by the FDA or the
European Medicines Agency, and to market, sell and distribute NeuVax in South Korea assuming such approval is obtained.
Recent Developments (in
reverse chronological order)
Litigation Settlement
On December 4, 2015, we reported that we had reached an agreement in principle to settle the consolidated shareholder derivative
action, captioned In re Galena Biopharma, Inc. Derivative Litigation, Civil Action No. 3:14-cv-00382-SI, currently pending in the United States District Court for the District of Oregon against the Company and/or certain of its
current and former officers and directors and the consolidated securities putative class action lawsuits, captioned In re Galena Biopharma, Inc. Securities Litigation, Civil Action No. 3:14-cv-00367-SI, pending against the
Company, certain of its current and former officers and directors, and other defendants in the United States District Court for the District of Oregon.
Strategic Review of Commercial Operations – We have completed a strategic review of our commercial operations.
During the three months ended September 30, 2015, we completed a strategic review of the commercial business and operations. As a result
of the review, we recently sold our Abstral® (fentanyl) Sublingual Tablets product and related assets, and we have determined to sell or otherwise dispose of our Zuplenz® (odansetron) Oral Soluble Film product and related assets and to cease our commercial operations.
GALE-302 Immunological Data Optimizing GALE-301 Presented - We presented preliminary immunologic data for our GALE-301 and
GALE-302 Phase 1b clinical trial at the Society for Immunotherapy of Cancer (SITC) 30th Anniversary Annual Meeting.
On November 7,
2015 we presented the poster, entitled, “Preliminary report of a clinical trial supporting the sequential use of an attenuated E39 peptide (E39’) to optimize the immunologic response to the FBP (E39+GM-CSF) vaccine,” that compared
three primary vaccine series (PVS) sequences of GALE-301 (E39) and GALE-302 (E39’) in ovarian and breast cancer patients to optimize the ex vivo immune responses, local reactions (LR), and delayed type hypersensitivity (DTH) reactions. The data
demonstrated that the in vivo immune response is enhanced with the use of the attenuated E39’ (GALE-302) after E39 (GALE-301). The optimal vaccination sequence utilizing three inoculations of GALE-301 followed by three inoculations of GALE-302
produced the most prominent and statistically significant LR and DTH responses.
Patent Infringement Litigation – We
have settled the patent infringement litigation involving Abstral.
On October 27, 2015, we reported that on October 23,
2015 we, Orexo AB and Actavis Laboratories Fl, Inc. (“Actavis”) had entered into a settlement and license agreement, which will permit Actavis to enter the market with a generic or authorized generic version of Abstral in June 2018 or
earlier under certain circumstances. The litigation, Orexo AB v. Actavis Laboratories FL, Inc., CA
7
Prospectus No. 1
No. 3:15-cv-00826, was dismissed with prejudice by the United States District Court for the District of New Jersey before and the agreement will be filed with the Federal Trade Commission
and the Department of Justice under applicable law.
Collaboration with the National Cancer Institute – We announced a
Phase 2 clinical trial with NeuVax in Ductal Carcinoma in Situ Patients.
On September 30, 2015, we announced a collaboration
with the National Cancer Institute (NCI) to initiate a Phase 2 clinical trial with NeuVax in patients diagnosed with Ductal Carcinoma in Situ (DCIS). The trial will be entitled VADIS: Phase 2 trial of the Nelipepimut-S Peptide Vaccine in Women with
DCIS of the Breast. The University of Texas M.D. Anderson Cancer Center (MDACC) Phase I and II Chemoprevention Consortium will be the lead clinical trial site for this multi-center trial with Elizabeth Mittendorf, M.D., Ph.D. serving as the
study Principal Investigator. The Consortium is funded through the Division of Cancer Prevention at NCI, which will provide financial and administrative support for the trial. We will provide NeuVax, as well as additional financial and
administrative support. The single-blind, double arm, randomized, controlled trial is expected to initiate in the fourth quarter of 2015/ first quarter of 2016.
GALE-301 Phase 2a Clinical Trial Data Presented – We announced positive data for our GALE-301 Phase 2a clinical trial
On September 28, 2015 we announced the poster presentation of positive data from the GALE-301 Phase 2a portion of the Phase
1/2a clinical trial at the European Cancer Congress 2015, providing updated data for all ovarian and endometrial cancer patients who have received at least twelve months of treatment on the study. The poster was entitled, entitled
“Preliminary results of the phase I/IIa dose finding trial of a folate binding protein vaccine GALE-301 (E39) + GM-CSF in ovarian and endometrial cancer patients to prevent recurrence,” and as presented, the clinical recurrence rate based
on all treatment cohorts was 41% in the Vaccine Group (VG) (n=29) versus 55% in the Control Group (CG) (n=22), p=0.41. However, in the 1000 mcg VG cohort (n=15), there have only been two clinical recurrences (13.3% versus 55% CG, p=0.02), and
the two-year Disease Free Survival (DFS) estimate is 85.7% (1000 mcg patients) versus 33.6% (CG), p < 0.02, as compared by Kaplan-Meir and Log rank tests.
IDMC Provides Recommendation to Reduce Cardiac Monitoring in the PRESENT Trial – We announced that the Independent Data
Monitoring Committee (IDMC) for the NeuVax Phase 3 clinical trial recommended to the Company that it can reduce the cardiac toxicity monitoring for patients in our PRESENT study
On August 24, 2015 we announced that the Independent Data Monitoring Committee (IDMC) for the NeuVax Phase 3 clinical trial recommended
to the Company that we can reduce the cardiac toxicity monitoring for patients in our PRESENT study. Following its most recent meeting in June 2015, the IDMC recommended routine cardiac monitoring could be reduced in the PRESENT trial and that such
a reduction is justified and consistent with the pre-specified Cardiac Toxicity Monitoring Stopping Rules defined in the study protocol. The IDMC concluded that cardiac toxicity monitoring by echocardiogram (ECHO) or multiple-gated acquisition
(MUGA) scans could be reduced. The IDMC had no other suggestions and recommended the trial continue as planned.
GALE 401 Phase 2
Clinical Trial Data Presented - We presented data from our Phase 2 clinical trial of GALE-401 at the European Hematology Association 20th Congress
On June 15, 2015, we announced our poster presentation entitled “Phase 2 Study of a Novel Controlled-Release Formulation of
Anagrelide (GALE-401) in Subjects with Myeloproliferative Neoplasm (MPN)-Related Thrombocytosis,” which was presented on June 13, 2015. The Phase 2 study demonstrated that GALE-401 was well tolerated with primarily Grade 1 and 2
toxicities in 16 of the 18 subjects enrolled. The efficacy shown in the trial compares favorably to historical anagrelide immediate release (IR) response rates with the following platelet response: overall response rate (ORR) of 78% (14/18);
complete response (CR) of 39% (7/18); partial response (PR) of 39% (7/18). The median time to response was 5 weeks (range, 3-10), and the median duration of response has not yet been reached.
Published Abstracts related to GALE-301 and Leica Biosystem’s Companion Diagnostic - We presented two abstract publications
at the American Society of Clinical Oncology (ASCO) 2015 Annual Meeting.
On May 27, 2015, we announced two abstract
publications at the ASCO 2015 Annual Meeting related to our cancer immunotherapy programs:
We presented data related to GALE-301 in
abstract #e14031, entitled, “Preliminary Results of the Phase I/IIa Dose Finding Trial of a Folate Binding Protein Vaccine (E39+GM-CSF) in Ovarian and Endometrial Cancer Patients to Prevent Recurrence.” The more mature data set from this
trial was presented in September 2015 as mentioned above.
8
Prospectus No. 1
We presented data related to our NeuVax Phase 3 PRESENT trial companion diagnostic in abstract
#e11609, entitled, “Analytical Validation of BOND Oracle HER2 IHC System for Identifying Low to Intermediate HER2 Expressing Breast Cancer in NeuVax PRESENT Phase 3 Clinical Trial.” This data demonstrated a direct correlation between cell
line receptor load, quantitative measure of HER2 protein, and IHC score. The ability to discriminate HER2 protein expression at the low and intermediate levels in breast cancer tumors will identify patients for new treatments in development such as
NeuVax. Specifically, the validation of the Bond Oracle HER2 IHC System to distinguish lower levels of HER2+ expressions supports its use as a companion diagnostic.
NeuVax Phase 3 PRESENT Over-Enrollment Completed - We announced the completion of enrollment in our NeuVax Phase 3 PRESENT
clinical trial.
On April 14, 2015, we announced the completion of enrollment in the NeuVax™ Phase 3 PRESENT clinical
trial. As anticipated, we over-enrolled the trial by 7.7% with a total of 758 patients now in the intent-to-treat (ITT) population. The protocol for the PRESENT trial called for 700 patients; and we expect this higher number of ITT patients will
increase the confidence in both the timing and quality of the statistics and the final outcome of the trial. The primary endpoint is currently expected to be reached in 2018, after the last patient dosed reaches her 36th month of treatment, or a
total of 141 events (recurrence or death) occur, whichever comes later.
Expanded Patient Population in NeuVax Combination
Trial - We announced the expansion of the patient population in the NeuVax and trastuzumab combination trial in HER2 1+/2+ patients to include HLA A24+ or HLA-A26+ patients.
On March 26, 2015, we announced that human leukocyte antigen (HLA) - A24+ or HLA-A26+ women are now eligible for enrollment into the
ongoing Phase 2b clinical trial with NeuVax in combination with trastuzumab. The trial evaluates node positive and triple negative, node negative breast cancer patients with immunohistochemistry (IHC) HER2 1+/2+ expressing tumors who are
disease-free after standard of care therapy.
Enrolled 700th Patient in NeuVax Phase 3 PRESENT Clinical Trial —We
announced enrollment of the 700th patient in our Phase 3 PRESENT clinical trial.
On February 9, 2015, we announced the
enrollment in the NeuVax Phase 3 PRESENT clinical trial of the 700th patient, which is the patient enrollment target as defined by the PRESENT Phase 3 clinical trial protocol.
Financial Condition
We had cash and cash
equivalents of approximately $34.8 million as of September 30, 2015. We believe that our existing cash and cash equivalents together with funding available under our purchase agreement with Lincoln Park Capital Fund, LLC and sales agreements
with MLV & Co. and Maxim Group LLC should be sufficient to fund our operations for at least one year. This projection is based on our current planned operations and revenue expectations and is subject to changes in our plans and
uncertainties inherent in our business, and we may need to seek to replenish our existing cash and cash equivalents sooner than we project. There is no guarantee that any additional funding will be available to us on acceptable terms, or at all. If
we fail to obtain additional funding when needed, we would be forced to scale back or terminate our operations, or to seek to merge with or to be acquired by another company.
Purchase Agreement with Lincoln Park Capital Fund, LLC
On November 18, 2014, we entered into a purchase agreement with Lincoln Park Capital Fund, LLC, or Lincoln Park. The purchase agreement
provides that in our discretion, upon the terms and subject to the conditions and limitations set forth therein, Lincoln Park currently is irrevocably committed to purchase from us an aggregate of up to $42,194,000 of additional shares of our common
stock over the remaining term of the purchase agreement.
There is no upper limit on the price per share that Lincoln Park must pay for
our common stock under the purchase agreement, but in no event will shares be sold to Lincoln Park on a day our closing price is less than $1.00 per share. The purchase price will be equitably adjusted for any reorganization, recapitalization,
non-cash dividend, stock split or other similar transaction occurring during the business days used to compute the purchase price.
There
are no trading volume requirements or restrictions under the purchase agreement, but there are limitations on the number of shares we can sell to Lincoln Park as described below. We will control the timing and amount of any sales of our common stock
to Lincoln Park, and we may terminate the purchase agreement at any time without fee, penalty or cost, upon one business day notice to Lincoln Park.
The purchase agreement limits our sales of shares of common stock to Lincoln Park to a maximum of 19.99% of our total outstanding shares of
common stock as of November 18, 2014, or approximately 42,270,277 shares, including 7,932,121 shares sold
9
Prospectus No. 1
to Lincoln Park prior to the date of this prospectus, unless we first obtain stockholder approval under the rules of The NASDAQ Capital Market or unless the average price of all shares of our
common stock sold to Lincoln Park exceeds $1.889, which represents the closing bid price of our common stock on November 18, 2014 as reported on The NASDAQ Capital Market plus $0.0491, such that such sales to Lincoln Park are considered to be
at least “at market” under the applicable NASDAQ rules.
The purchase agreement also prohibits us from selling Lincoln Park any
shares of common stock if those shares, when aggregated with all other shares of our common stock then beneficially owned by Lincoln Park and its affiliates, would result in Lincoln Park and its affiliates having beneficial ownership of more than
9.99% of the then total outstanding shares of our common stock (approximately 16,174,000 shares as of September 30, 2015), as calculated pursuant to Section 13(d) of the Securities Exchange Act of 1934, as amended, or the Exchange Act, and
Rule 13d-3 thereunder.
Sales Agreements with MLV & Co. and Maxim Group LLC
We entered into sales agreements with MLV & Co. LLC, or MLV, and Maxim Group LLC, or Maxim, on May 24, 2013, under which we may
currently issue and sell our common stock having a cumulative aggregate offering price of up to $20,000,000.
The sales, if any, of shares
made under the sales agreements will be made on The NASDAQ Capital Market by means of ordinary brokers’ transactions at market prices, in block transactions or as otherwise agreed by MLV or Maxim and us. We may instruct MLV and Maxim not to
sell common stock if the sales cannot be effected at or above the price designated by us from time to time. We or MLV or Maxim may suspend the offering of common stock upon notice and subject to other conditions. As agents, MLV and Maxim will not
engage in any transactions that stabilize the price of our common stock.
We will pay each of MLV and Maxim commissions for its services
in acting as agent in the sale of common stock. MLV and Maxim will be entitled to compensation at a fixed commission rate of 3.0% of the gross sales price per share sold.
Corporate Information
Our principal
executive offices are located at 2000 Crow Canyon Place, Suite 380, San Ramon, California 94583, and our phone number is (855) 855-4253. Our website address is www.galenabiopharma.com. We do not incorporate the information on our website into
this prospectus, and you should not consider such information part of this prospectus.
We were incorporated as Argonaut Pharmaceuticals,
Inc. in Delaware on April 3, 2006 and changed our name to RXi Pharmaceuticals Corporation on November 28, 2006. On September 26, 2011, we changed our company name from RXi Pharmaceuticals Corporation to Galena Biopharma, Inc.
USE OF PROCEEDS
Unless we state otherwise in the accompanying prospectus supplement, we currently intend to use the net proceeds from this offering to augment
our working capital and for general corporate purposes, including our Phase 3 PRESENT study and other clinical trials of our product candidates. General corporate purposes also may include, but are not limited to, payments relating to the
ongoing defense or settlement of pending stockholder lawsuits or the possible sale or other disposition of our marketed products and discontinuation of our commercial operations, repayment of indebtedness, financing of capital expenditures and
potential future acquisitions and strategic investments. Pending application of the net proceeds, we expect to invest the net proceeds in investment-grade, interest-bearing securities.
10
Prospectus No. 1
FINANCIAL RATIOS
We present below the ratio of our earnings to combined fixed charges and preference dividends, if any. Earnings consist of our net loss plus
fixed charges. Fixed charges consist of interest expense and amortization of debt issuance costs. We recorded no preference dividends in any of the periods presented.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| |
|
Year Ended December 31, |
|
|
Nine
Months
Ended
September
30, |
|
| |
|
2010 |
|
|
2011 |
|
|
2012 |
|
|
2013 |
|
|
2014 |
|
|
2015 |
|
| Ratio of earnings (loss) to combined fixed charges and preference dividends(1) |
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
— |
|
|
|
|
|
|
|
|
| Deficiency in earnings (loss) to cover fixed charges |
|
|
N/A |
|
|
|
N/A |
|
|
|
N/A |
|
|
|
N/A |
|
|
|
N/A |
|
|
|
N/A |
|
| (1) |
We did not record earnings in any of the periods presented. Accordingly, our earnings were insufficient to cover fixed charges in such periods, and we are unable to disclose a ratio of earnings to combined fixed charges
and preference dividends for such periods. |
THE SECURITIES THAT WE MAY OFFER
We, directly or through agents, dealers or underwriters designated from time to time, may offer, issue and sell, together or separately, up to
$100,000,000 in the aggregate of:
| |
• |
|
shares of our common stock, par value $0.0001 per share; |
| |
• |
|
shares of our preferred stock, par value $0.0001 per share; |
| |
• |
|
warrants to purchase our common stock or preferred stock; |
| |
• |
|
rights to purchase our common stock or preferred stock; and |
| |
• |
|
any combination of the securities listed above, separately or as units, each on terms to be determined at the time of sale. |
The common stock, preferred stock, warrants, rights and units collectively are referred to in this prospectus as the “securities.”
We have summarized below the material terms of the various types of securities that we may offer. We will describe in the applicable
prospectus supplement the detailed terms of the securities offered by that supplement. If indicated in the prospectus supplement, the terms of the offered securities may differ from the terms summarized below.
DESCRIPTION OF CAPITAL STOCK
General
Our authorized capital stock
consists of 280,000,000 shares, all which includes:
| |
• |
|
275,000,000 shares of common stock, par value $0.0001 per share, and |
| |
• |
|
5,000,000 shares of preferred stock, par value $0.0001 per share. |
As of September 30,
2015, there were 161,900,446 shares of common stock outstanding held by approximately 544 stockholders of record, and no shares of preferred stock outstanding. The number of holders of record does not include shares held in “street name”
through brokers.
11
Prospectus No. 1
Common Stock
Each holder of our common stock is entitled to one vote for each share of common stock held on all matters submitted to a vote of stockholders.
Common stockholders are not entitled to cumulative voting in the election of directors by our certificate of incorporation. This means that the holders of a majority of the shares voted will be able to elect all of the directors then standing for
election. Subject to preferences that may apply to shares of preferred stock outstanding at the time, the holders of outstanding shares of our common stock are entitled to receive dividends out of assets legally available at the times and in the
amounts that our board of directors may determine from time to time.
Upon our liquidation, dissolution or winding-up, the holders of our
common stock will be entitled to share ratably in all assets remaining after payment of all liabilities and the liquidation preferences of any series of capital stock ranking senior to the common stock upon liquidation. Holders of common stock have
no preemptive or conversion rights or other subscription rights. There will be no redemption or sinking fund provisions applicable to the common stock. All outstanding shares of common stock are fully paid and nonassessable, and the shares of common
stock to be issued under this prospectus, when they are paid for, will be fully paid and nonassessable.
Preferred Stock
Our board of directors is authorized, subject to any limitations prescribed by law, without further vote or action by the stockholders, to
issue from time to time any of the authorized shares of preferred stock in one or more series without stockholder approval. Each such series of preferred stock shall have such number of shares, designations, preferences, voting powers,
qualifications, and special or relative rights or privileges as shall be determined by our board of directors, which may include, among others, dividend rights, voting rights, liquidation preferences, conversion rights and preemptive rights.
Our board of directors will fix the rights, preferences, privileges, qualifications and restrictions of the preferred stock of each series
that we sell under this prospectus in the certificate of designation relating to each such series. We will incorporate by reference as an exhibit to the registration statement of which this prospectus is a part or as an exhibit to one or more
current reports on Form 8-K, the form of any certificate of designation that describes the terms of the series of preferred stock we are offering before the issuance of the related series of preferred
stock. This description will include:
| |
• |
|
the title and stated value; |
| |
• |
|
the number of shares we are offering; |
| |
• |
|
the liquidation preference per share; |
| |
• |
|
the purchase price per share; |
| |
• |
|
the dividend rate per share, dividend period, payment date or dates and method of calculation of dividends; |
| |
• |
|
whether dividends will be cumulative or non-cumulative and, if cumulative, the date from which dividends will accumulate; |
| |
• |
|
our right, if any, to defer payment of dividends and the maximum length of any such deferral period; |
| |
• |
|
the procedures for any auction and remarketing, if any; |
| |
• |
|
the provisions for a sinking fund, if any; |
| |
• |
|
the provisions for redemption or repurchase, if applicable, and any restrictions on our ability to exercise those redemption and repurchase rights; |
| |
• |
|
any listing of the preferred stock on any securities exchange or market; |
| |
• |
|
whether the preferred stock will be convertible into our common stock or other securities of ours, including warrants, and, if applicable, the conversion price, or how it will be calculated, and under what circumstances
and the mechanism by which it may be adjusted, and the conversion period; |
| |
• |
|
whether the preferred stock will be exchangeable into debt securities or other securities of ours, and, if applicable, the exchange price, or how it will be calculated, and under what circumstances it may be adjusted,
and the exchange period; |
| |
• |
|
preemptive rights, if any; |
12
Prospectus No. 1
| |
• |
|
restrictions on transfer, sale or other assignment, if any; |
| |
• |
|
a discussion of any material United States federal income tax considerations applicable to the preferred stock; |
| |
• |
|
the relative ranking and preferences of the preferred stock as to dividend rights and rights if we liquidate, dissolve or wind up our affairs; |
| |
• |
|
any limitations on issuances of any class or series of preferred stock ranking senior or on a parity with the series of preferred stock being issued as to dividend rights and rights if we liquidate, dissolve or wind up
our affairs; and |
| |
• |
|
any other specific terms, rights, preferences, privileges, qualifications or limitations of, or restrictions on, the preferred stock. |
If we issue and sell shares of preferred stock pursuant to this prospectus, the shares will be fully paid and nonassessable and will not have,
or be subject to, any preemptive or similar rights.
The laws of the State of Delaware, the state of our incorporation, provide that the
holders of preferred stock will have the right to vote separately as a class on any proposal involving fundamental changes in the rights of holders of such preferred stock. This right is in addition to any voting rights that may be provided for in
the applicable certificate of designation.
We believe the power to issue preferred stock will provide our board of directors with
flexibility in connection with certain possible corporate transactions. The issuance of preferred stock, however, could adversely affect the voting power of holders of our common stock, restrict their rights to receive payment upon liquidation, and
have the effect of delaying, deferring, or preventing a change in control which may be beneficial to our stockholders.
Anti-Takeover Effects of
Delaware Law and Our Restated Certificate of Incorporation and Bylaws
Certain provisions of Delaware law, our amended and restated
certificate of incorporation and our amended and restated bylaws contain provisions that could have the effect of delaying, deferring or discouraging another party from acquiring control of us. These provisions, which are summarized below, are
expected to discourage certain types of coercive takeover practices and inadequate takeover bids. These provisions are also designed, in part, to encourage persons seeking to acquire control of us to first negotiate with our board of directors. We
believe that the benefits of increased protection of our potential ability to negotiate with an unfriendly or unsolicited acquirer outweigh the disadvantages of discouraging such proposals, including proposals that are priced above the then-current
market value of our common stock, because, among other reasons, the negotiation of such proposals could result in an improvement of their terms.
Certificate of Incorporation and Bylaws
Our amended and restated certificate of incorporation and amended and restated bylaws include provisions that:
| |
• |
|
authorize our board of directors to issue, without further action by the stockholders, up to 5,000,000 shares of undesignated preferred stock; |
| |
• |
|
require that any action to be taken by our stockholders be effected at a duly called annual or special meeting and not by written consent; |
| |
• |
|
specify that special meetings of our stockholders can be called only by our board of directors, the chairman of the board or the chief executive officer; |
| |
• |
|
provide that our board of directors will be classified, with directors serving staggered three-year terms; |
| |
• |
|
provide that directors may be removed only for cause and may only be removed for cause only by the holders of 75% of our outstanding capital stock entitled to vote generally in the election of directors;
|
| |
• |
|
provide that vacancies on our board of directors may be filled only by a majority of directors then in office, even though less than a quorum; |
| |
• |
|
provide for a 75% vote of stockholders to amend our amended and restated bylaws, unless the amendment has been approved by a majority of our directors who are not affiliated or associated with any person or entity
holding 10% or more of the voting power of our outstanding capital stock; and |
13
Prospectus No. 1
| |
• |
|
establish an advance notice procedure for stockholder proposals to be brought before an annual meeting of our stockholders, including proposed nominations of persons for election to our board of directors.
|
Delaware Anti-Takeover Statute
We are subject to the provisions of Section 203 of the Delaware General Corporation Law regulating corporate takeovers. In general,
Section 203 prohibits a publicly-held Delaware corporation from engaging, under certain circumstances, in a business combination with an interested stockholder for a period of three years following the date the person became an interested
stockholder unless:
| |
• |
|
prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder;
|
| |
• |
|
upon completion of the transaction that resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the
transaction commenced, excluding for purposes of determining the voting stock outstanding, but not the outstanding voting stock owned by the interested stockholder, (1) shares owned by persons who are directors and also officers and
(2) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| |
• |
|
at or subsequent to the date of the transaction, the business combination is approved by the board of directors of the corporation and authorized at an annual or special meeting of stockholders, and not by written
consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
Generally, a “business combination” includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to
the “interested stockholder.” Generally, an “interested stockholder” is a person who, together with affiliates and associates, owns or, within three years prior to the determination of interested stockholder status, did own 15%
or more of a corporation’s outstanding voting stock. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that
Section 203 may discourage business combinations or other attempts that might result in a premium over the market price for the shares of common stock held by our stockholders.
The provisions of Delaware law, our restated certificate of incorporation and our amended and restated bylaws could have the effect of
discouraging others from attempting hostile takeovers and, as a consequence, they may also inhibit temporary fluctuations in the market price of our common stock that often result from actual or rumored hostile takeover attempts. These provisions
may also have the effect of preventing changes in our management. It is possible that these provisions could make it more difficult to accomplish transactions that stockholders may otherwise deem to be in their best interests.
Listing
Our common stock is listed on
The NASDAQ Capital Market under the symbol “GALE.”
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Computershare Trust Company, N.A.
DESCRIPTION OF WARRANTS
We may issue warrants for the purchase of preferred stock or common stock, or a combination thereof. We may issue warrants independently or
together with any other securities offered by any prospectus and may be attached to or separate from the other offered securities. Each series of warrants will be issued under a separate warrant agreement to be entered into by us with a warrant
agent. The warrant agent will act solely as our agent in connection with the warrants and will not assume any obligation or relationship of agency or trust for or with any holders or beneficial owners of warrants. Further terms of the warrants and
the applicable warrant agreements will be set forth in the applicable prospectus supplement.
The prospectus supplement relating to any
particular issue of warrants will describe the terms of the warrants, including, as applicable, the following:
| |
• |
|
the title of the warrants; |
14
Prospectus No. 1
| |
• |
|
the aggregate number of the warrants; |
| |
• |
|
the price or prices at which the warrants will be issued; |
| |
• |
|
the designation, terms and number of shares of debt securities, preferred stock or common stock purchasable upon exercise of the warrants; |
| |
• |
|
the designation and terms of the offered securities, if any, with which the warrants are issued and the number of the warrants issued with each offered security; |
| |
• |
|
the date, if any, on and after which the warrants and the related debt securities, preferred stock or common stock will be separately transferable; |
| |
• |
|
the price at which each share of debt securities, preferred stock or common stock purchasable upon exercise of the warrants may be purchased; |
| |
• |
|
the date on which the right to exercise the warrants shall commence and the date on which that right shall expire; |
| |
• |
|
the minimum or maximum amount of the warrants which may be exercised at any one time; |
| |
• |
|
information with respect to book-entry procedures, if any; |
| |
• |
|
a discussion of certain federal income tax considerations; and |
| |
• |
|
any other terms of the warrants, including terms, procedures and limitations relating to the exchange and exercise of the warrants. |
We and the warrant agent may amend or supplement the warrant agreement for a series of warrants without the consent of the holders of the
warrants issued thereunder to effect changes that are not inconsistent with the provisions of the warrants and that do not materially and adversely affect the interests of the holders of the warrants.
DESCRIPTION OF RIGHTS
We may issue rights to purchase common stock or preferred stock. The accompanying prospectus supplement will contain the material terms and
conditions for each right.
We will describe in the applicable prospectus supplement the terms and conditions of the issue of rights being
offered, the rights agreement relating to the rights and the rights certificates representing the rights, including, as applicable:
| |
• |
|
the title of the rights; |
| |
• |
|
the date of determining the stockholders entitled to the rights distribution; |
| |
• |
|
the title, aggregate number of shares of common stock or preferred stock purchasable upon exercise of the rights; |
| |
• |
|
the aggregate number of rights issued; |
| |
• |
|
the date, if any, on and after which the rights will be separately transferable; |
| |
• |
|
the date on which the right to exercise the rights will commence and the date on which the right will expire; and |
| |
• |
|
any other terms of the rights, including terms, procedures and limitations relating to the distribution, exchange and exercise of the rights. |
Each right will entitle the holder of rights to purchase for cash the principal amount of shares of common stock or preferred stock at the
exercise price provided in the applicable prospectus supplement. Rights may be exercised at any time up to the close of business on the expiration date for the rights provided in the applicable prospectus supplement. After the close of business on
the expiration date, all unexercised rights will be void.
15
Prospectus No. 1
Holders may exercise rights as described in the applicable prospectus supplement. Upon receipt of
payment and the rights certificate properly completed and duly executed at the corporate trust office of the rights agent or any other office indicated in the prospectus supplement, we will, as soon as practicable, forward the shares of common stock
or preferred stock purchasable upon exercise of the rights. If less than all of the rights issued in any rights offering are exercised, we may offer any unsubscribed securities directly to persons other than stockholders, to or through agents,
underwriters or dealers or through a combination of such methods, including pursuant to standby underwriting arrangements, as described in the applicable prospectus supplement.
DESCRIPTION OF UNITS
We may offer and issue units that consist of shares of our common stock or preferred stock and warrants to purchase additional shares of our
common stock or preferred stock. For example, we may elect to issue units for a specified price per unit, with each unit consisting of one share of our common stock or preferred stock and one warrant to purchase an additional share of our common
stock or preferred stock at a specified price. The holder of a unit will also hold each of the securities that is included in the unit.
We have provided in the preceding sections of this prospectus a general description of our common stock, preferred stock, and warrants that we
may offer. If we elect to offer units, we will describe the specific terms of the units in a supplement to this prospectus. Among other things, the prospectus supplement will describe, to the extent applicable:
| |
• |
|
the price of each unit; |
| |
• |
|
the securities comprising each unit; |
| |
• |
|
the exercise price of the warrants comprising part of the units; |
| |
• |
|
the aggregate number of units offered; |
| |
• |
|
the number of shares of common stock or preferred stock that may be purchased upon the exercise of each warrant comprising part of a unit; |
| |
• |
|
the terms of any right by us to redeem any of the securities comprising the units; |
| |
• |
|
the date on which the right to exercise the warrants forming part of the units will commence and the date on which this right will expire; |
| |
• |
|
any transfer restrictions on the units, including whether the securities comprising the units may be transferred separately; |
| |
• |
|
the terms on which the units or warrants forming part of the units may be amended; |
| |
• |
|
with respect to preferred stock forming part of the units, the other matters listed above under “Description of Capital Stock—Preferred Stock”; |
| |
• |
|
with respect to warrants forming part of the units, the other matters listed above under “Description of Warrants”; and |
| |
• |
|
the material United States federal income tax consequences applicable to the units. |
PLAN OF DISTRIBUTION
We may sell the securities offered by this prospectus to one or more underwriters or dealers for public
offering and sale by them or to investors directly or through agents. The accompanying prospectus supplement will set forth the terms of the offering and the method of distribution and will identify any firms acting as underwriters, dealers or
agents in connection with the offering, including:
| |
• |
|
the name or names of any underwriters, dealers or agents; |
| |
• |
|
the purchase price of the securities and the proceeds to us or to any selling stockholder from the sale; |
| |
• |
|
any underwriting discounts and other items constituting compensation to underwriters, dealers or agents; |
| |
• |
|
any public offering price; |
16
Prospectus No. 1
| |
• |
|
any discounts or concessions allowed or reallowed or paid to dealers; and |
| |
• |
|
any securities exchange or market on which the securities offered in the prospectus supplement may be listed. |
Only those underwriters identified in such prospectus supplement are deemed to be underwriters in connection with the securities offered in
the prospectus supplement.
The distribution of the securities may be effected from time to time in one or more transactions at a fixed
price or prices, which may be changed, or at prices determined as the applicable prospectus supplement specifies. The securities may be sold through a rights offering, forward contracts or similar arrangements. In connection with the sale of the
securities, underwriters, dealers or agents may be deemed to have received compensation from us or selling stockholders in the form of underwriting discounts or commissions and also may receive commissions from securities purchasers for whom they
may act as agent. Underwriters may sell the securities to or through dealers, and the dealers may receive compensation in the form of discounts, concessions or commissions from the underwriters or commissions from the purchasers for whom they may
act as agent. Some of the underwriters, dealers or agents who participate in the securities distribution may engage in other transactions with, and perform other services for, us in the ordinary course of business.
We will provide in the applicable prospectus supplement information regarding any underwriting discounts or other compensation that we pay to
underwriters or agents in connection with the securities offering, and any discounts, concessions or commissions which underwriters allow to dealers. Underwriters, dealers and agents participating in the securities distribution may be deemed to be
underwriters, and any discounts and commissions they receive and any profit they realize on the resale of the securities may be deemed to be underwriting discounts and commissions under the Securities Act of 1933. Underwriters and their controlling
persons, dealers and agents may be entitled, under agreements entered into with us, to indemnification against and contribution toward specific civil liabilities, including liabilities under the Securities Act.
The securities may or may not be listed on a national securities exchange. In connection with an offering, the underwriters may purchase and
sell securities in the open market. These transactions may include short sales, stabilizing transactions and purchases to cover positions created by short sales. Short sales involve the sale by the underwriters of a greater number of securities than
they are required to purchase in an offering. Stabilizing transactions consist of bids or purchases made for the purpose of preventing or retarding a decline in the market price of the securities while an offering is in progress. The underwriters
also may impose a penalty bid. This occurs when a particular underwriter repays to the underwriters a portion of the underwriting discount received by it because the underwriters have repurchased securities sold by or for the account of that
underwriter in stabilizing or short-covering transactions. These activities by the underwriters may stabilize, maintain or otherwise affect the market price of the securities. As a result, the price of the securities may be higher than the price
that otherwise might exist in the open market. If these activities are commenced, they may be discontinued by the underwriters at any time.
Any underwriter who is qualified market maker on The Nasdaq Capital Market may engage in passive market making transactions in the securities
on The Nasdaq Capital Market in accordance with Rule 103 of Regulation M, during the business day prior to the pricing of the offering, before the commencement of offers or sales of the securities. Passive market makers must comply with applicable
volume and price limitations and must be identified as passive market makers. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for such security; if all independent bids are lowered
below the passive market maker’s bid, however, the passive market maker’s bid must then be lowered when certain purchase limits are exceeded.
Certain underwriters, dealers or agents and their associates may engage in transactions with and perform services for us in the ordinary
course of our business.
LEGAL MATTERS
TroyGould PC, Los Angeles, California, has rendered an opinion with respect to the securities offered by this prospectus. Sanford J.
Hillsberg, the Chairman of our company, is an attorney with TroyGould PC. TroyGould PC owned 63,941 shares of our common stock as of September 30, 2015.
EXPERTS
The consolidated financial statements of the Company as of December 31, 2014 and 2013, and for the years then ended, and the effectiveness of
internal control over financial reporting of the Company as of December 31, 2014, have been incorporated by reference herein in reliance upon the reports of Moss Adams LLP, independent registered public accounting firm, incorporated by reference
herein, and upon the authority of said firm as experts in accounting and auditing.
17
Prospectus No. 1
The consolidated financial statements of Galena Biopharma, Inc. for the year ended
December 31, 2012 incorporated by reference in this prospectus have been so incorporated in reliance on the report of BDO USA, LLP, an independent registered public accounting firm, incorporated herein by reference, given on the authority of
said firm as experts in auditing and accounting.
WHERE YOU CAN FIND MORE INFORMATION
We have filed a registration statement on Form S-3 with the SEC under the Securities Act of 1933. This prospectus is part of the registration
statement but the registration statement includes and incorporates by reference additional information and exhibits. We file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and copy the
registration statement and any other document we file with the SEC at the public reference room maintained by the SEC at 100 F Street, N.E., Washington, D.C. 20549. You may obtain information on the operation of the public reference room by calling
the SEC at 1-800-SEC-0330. The SEC also maintains a web site that contains reports, proxy and information statements and other information regarding companies, such as ours, that file documents electronically with the SEC. The address of that site
on the world wide web is http://www.sec.gov. The information on the SEC’s web site is not part of this prospectus, and any references to this web site or any other web site are inactive textual references only.
INCORPORATION OF CERTAIN DOCUMENTS BY REFERENCE
The SEC allows us to “incorporate by reference” information from other documents that we file with it, which means that we can
disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this prospectus. Information in this prospectus supersedes information incorporated by reference that we
filed with the SEC prior to the date of this prospectus, while information that we file later with the SEC will automatically update and supersede the information in this prospectus. We incorporate by reference into this prospectus and the
registration statement of which this prospectus is a part the information or documents listed below that we have filed with the SEC:
| |
• |
|
our Annual Report on Form 10-K for the year ended December 31, 2014, which was filed on March 5, 2015, and amended on March 10, 2015 (Items 6, 7, and 8 from said Annual Report have been recast for discontinued
operations as reflected in the Current Report on Form 8-K filed on December 4, 2015); |
| |
• |
|
our Quarterly Report on Form 10-Q for the three months ended March 31, 2015 filed with the SEC on May 7, 2015; |
| |
• |
|
our Quarterly Report on Form 10-Q for the three months ended June 30, 2015 filed with the SEC on August 6, 2015; |
| |
• |
|
our Quarterly Report on Form 10-Q for the three months ended September 30, 2015 filed with the SEC on November 9, 2015; |
| |
• |
|
our Current Reports on Form 8-K filed with the SEC on January 14, 2015, February 9, 2015, March 5, 2015, March 16, 2015, April 9, 2015, May 7, 2015, June 24, 2015, August 6, 2015, October 27, 2015, October 30, 2015,
November 20, 2015, and December 4, 2015, respectively; and |
| |
• |
|
the description of our common stock contained in our registration statement on Form 8-A (File No. 001-33958) filed pursuant to Section 12 of the Exchange Act, as amended from time to time. |
We also incorporate by reference into this prospectus all documents filed by us with the SEC pursuant to Sections 13(a), 13(c), 14
or 15(d) of the Securities Exchange Act of 1934 prior to the termination of this offering, including all such documents that we may file with the SEC after the date of the initial registration statement and prior to the effectiveness of the
registration statement, except that we are not incorporating any information furnished under Item 2.02 or Item 7.01 of any Current Report on Form 8-K we may subsequently file.
You may request a copy of the documents incorporated by reference into this prospectus, at no cost, by writing or telephoning us at the
following address:
Galena Biopharma, Inc.
2000 Crow Canyon Place, Suite 380
San Ramon, California 94583
Attention: Investor Relations
Phone: (855) 855-4253
Copies of these documents are also available, without charge, through the “Investor Relations” section of our website
(www.galenabiopharma.com) as soon as reasonably practicable after it are filed with the SEC. The information contained on our website is not a part of this prospectus.
18
Prospectus No. 1
19,772,727 Shares of Common Stock
|
|
|
| Warrants to Purchase 11,863,636 Shares of Common Stock |

PROSPECTUS SUPPLEMENT
RAYMOND JAMES
|
|
|
|
|
|
|
|
|
|
|
| Roth Capital Partners |
|
|
Noble Financial Capital Markets |
|
|
|
FBR |
|
|
Maxim Group LLC |
January 7, 2016
SELLAS Life Sciences (NASDAQ:SLS)
Historical Stock Chart
From Mar 2024 to Apr 2024

SELLAS Life Sciences (NASDAQ:SLS)
Historical Stock Chart
From Apr 2023 to Apr 2024
